 Jörg Kumbrink1,2*
Jörg Kumbrink1,2* Melanie-Christin Demes3Jan Jeroch3Andreas Bräuninger4Kristin Hartung4Uwe Gerstenmaier5Ralf Marienfeld5Axel Hillmer6Nadine Bohn7Christina Lehning7Ferdinand Ferch7Peter Wild3Stefan Gattenlöhner4Peter Möller5Frederick Klauschen1,2Andreas Jung1,2
Melanie-Christin Demes3Jan Jeroch3Andreas Bräuninger4Kristin Hartung4Uwe Gerstenmaier5Ralf Marienfeld5Axel Hillmer6Nadine Bohn7Christina Lehning7Ferdinand Ferch7Peter Wild3Stefan Gattenlöhner4Peter Möller5Frederick Klauschen1,2Andreas Jung1,2- 1Institute of Pathology, Faculty of Medicine, Ludwig Maximilian University of Munich (LMU), Munich, Germany
- 2German Cancer Consortium (DKTK), Partner Site Munich, Munich, Germany
- 3Dr. Senckenberg Institute of Pathology, University Hospital Frankfurt, Frankfurt, Germany
- 4Institute of Pathology, Justus Liebig University Giessen, Giessen, Germany
- 5Institute of Pathology, University Ulm, Ulm, Germany
- 6Institute of Pathology, University Hospital Cologne, Cologne, Germany
- 7Archer, Boulder, CO, United States
Lung cancer is a paradigm for a genetically driven tumor. A variety of drugs were developed targeting specific biomarkers requiring testing for tumor genetic alterations in relevant biomarkers. Different next-generation sequencing technologies are available for library generation: 1) anchored multiplex-, 2) amplicon based- and 3) hybrid capture-based-PCR. Anchored multiplex PCR-based sequencing was investigated for routine molecular testing within the national Network Genomic Medicine Lung Cancer (nNGM). Four centers applied the anchored multiplex ArcherDX-Variantplex nNGMv2 panel to re-analyze samples pre-tested during routine diagnostics. Data analyses were performed by each center and compiled centrally according to study design. Pre-defined standards were utilized, and panel sensitivity was determined by dilution experiments. nNGMv2 panel sequencing was successful in 98.9% of the samples (N = 90). With default filter settings, all but two potential MET exon 14 skipping variants were identified at similar allele frequencies. Both MET variants were found with an adapted calling filter. Three additional variants (KEAP1, STK11, TP53) were called that were not identified in pre-testing analyses. Only total DNA amount but not a qPCR-based DNA quality score correlated with average coverage. Analysis was successful with a DNA input as low as 6.25 ng. Anchored multiplex PCR-based sequencing (nNGMv2) and a sophisticated user-friendly Archer-Analysis pipeline is a robust and specific technology to detect tumor genetic mutations for precision medicine of lung cancer patients.
Introduction
Lung cancer, specifically non-small cell lung cancer (NSCLC), covering the large entity of adenocarcinomas of the lung, is a paradigm for precision medicine as there are several tumor drivers known for which targeted drugs have been developed, tested and approved by legal bodies, e.g., EMA (European Medicines Agency), FDA (Food and Drug Administration) and many others [1, 2]. In precision medicine, therapies are patient tailored (personalized medicine) depending on the genetic make-up of the tumor which has to be determined by molecular diagnostics also known as companion diagnostic or theranostic in this context [3–5]. Due to the many known tumor genetic alterations found in lung cancer on the one hand and low amounts of tissue (biopsy, aspirates) on the other hand it is of great advantage to run molecular pathological analyses using multiplexing systems. Thus, NGS (next-generation sequencing) is a good choice and therefore used by many laboratories [6].
In Germany, the national consortium nNGM (national Network Genomic Medicine lung cancer) was established to provide central high quality and continuously advanced diagnostic testing of lung cancer patients from university hospitals, specialized lung clinics and local practices by more than 20 regional university pathology centers. This structure warrants both expertise and up to date molecular pathological diagnostics for the daily care of lung cancer patients [7].
The nNGM follows the strategy to provide a cost-effective analysis of genes/biomarkers that are associated with therapeutic options (EMA approved drugs) or are investigated in German clinical trials which can be reimbursed by German health insurances. However, the reimbursement contract hardly covers the costs for utilization of larger NGS panels. Therefore, a small panel covering mutation hotspots/biomarkers in NSCLC was designed.
The nNGM algorithm includes besides immunostaining of certain markers (e.g., PD-L1) the parallel analyses for single and small multi nucleotide variants on DNA level as well as testing for gene fusions/translocations. Currently, the nNGM guidelines require on the one hand the NGS analysis of pre-defined genomic regions within 26 genes on DNA level. However, the NGS technology of choice is not mandatory. On the other hand, the method of choice for gene fusion/translocation/isoform analyses (ALK, MET exon 14 skipping, NTRK1/2/3, ROS1, RET) is not stipulated, accepting results from various validated methods including FISH (fluorescence in situ hybridization) and NGS (DNA- or RNA-based).
Since different methods, besides NGS, are allowed for gene fusion determination, here we only focus on the cost-effective NGS nNGM DNA panel for the determination of single nucleotide variants, insertions/deletions, duplications and splice site variants. The second version of the nNGM panel (nNGMv2) comprises: ALK, BRAF, CTNNB1, EGFR, ERBB2, FGFR1, FGFR2, FGFR3, FGFR4, HRAS, IDH1, IDH2, KEAP1, KRAS, MAP2K1, MET, NRAS, NTRK1, NTRK2, NTRK3, PIK3CA, PTEN, RET, ROS1, STK11 and TP53.
Different NGS technologies are available for library generation: 1) anchored multiplex- (AMP), 2) amplicon based- (AMPL) and 3) hybrid capture-based (HCP) -PCR [8, 9].
Archer’s VariantPlex technology is based on AMP and has several advantages including low required DNA-amount, high specificity, fast hands-on protocol and easy parallel sample-handling [9]. Moreover, a free sophisticated analysis pipeline is available (Archer Analysis suite, Archer DX). AMP utilizes primer extension and represents an efficient way for specific amplification of DNA (either genomic or cDNA). This technique takes advantage of two different gene specific primers (GSP1, GSP2) per target and allows the amplification of DNA of low quality and or at low amounts [9].
The aim of this multiccentric study was to establish and validate an AMP based cost-efficient NGS panel (ArcherDX VariantPlex nNGMv2), fulfilling the minimum requirements of the nNGM. Therefore, a panel was defined by the nNGM, subsequently designed by ArcherDX and finally tested and validated in a joint effort of four nNGM centers.
Materials and methods
Study design and samples
The study design is depicted in Figure 1. Briefly, the German Institutes of Pathology of the Goethe University (Frankfurt)—center CA, Justus-Liebig University Giessen (Giessen)—center CB, Ludwig-Maximilians-University (Munich)—center CC and University of Ulm (Ulm)—center CD participated in this study. Each center selected 21 to 26 DNA samples (in total 93 samples) from its archives that had been previously analyzed during routine diagnostics by panel sequencing/NGS. DNA had been isolated from formalin fixed paraffin embedded (FFPE) NSCLC tumor samples. The samples were selected to 1) encompass a large repertoire of different tumor genetic variants [SNV (single nucleotide variants); ins (insertions); del (deletions); delins, dup (duplications); splice site variants; rare variants] relevant in NSCLC treatment, 2) comprise a spectrum of low to high DNA concentrations (1–392 ng/μL, median = 23 ng/μL) as well as 3) to cover a broad range of tumor cell contents (2%–90%, median = 50%). The same DNA samples which had been used for the initial routine diagnostics were enrolled in the present validation of our custom ArcherDX VariantPlex nNGMv2 panel (for simplicity mentioned from now on as nNGMv2 panel, ArcherDX, Boulder, CO, United States). In addition, two patient samples (CC-02-28 and CC-03-29) were utilized for serial DNA dilution/reduction experiments for investigating sensitivity and reproducibility. Finally, the commercial Horizon OncoSpan FFPE control (Catalog ID: HD832; Horizon Discovery Ltd., Cambridge, United Kingdom) containing 26 variants verified by NGS and digital droplet PCR of which 13 were covered by our custom panel were analyzed by two centers.
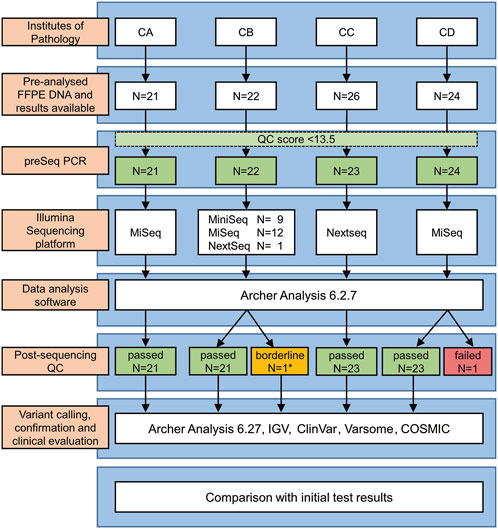
Figure 1. Study design. *Low leftover DNA volume (∼1/3 of the recommended volume).
Histopathological samples and DNA extraction
Leftover DNA isolated from tissue samples of NSCLC patients was used after completion of all diagnostic analyses. Originally, sections from FFPE tissue samples were prepared followed by hematoxylin-eosin (H&E) staining of a representative slide. Areas containing appropriate densities of tumor cells (Supplementary Table S1) were defined and marked by expert pathologists and used as blueprints for transferring marked areas onto unstained serial tissue sections. Marked areas were microdissected under microscopic control and genomic DNA was subsequently isolated using QIAamp DNA Micro Kits (Qiagen, Hilden, Germany; Center CA), Maxwell RSC FFPE Plus DNA Kits (Promega, Madison, WI, United States; Center CB), GeneRead DNA FFPE Kits (Qiagen; Center CC) or Qiagen FFPE AllPrep Kits (Qiagen; Center CD) according to the respective manufacturer’s instructions. DNA concentrations were measured using Qubit3 (Centers CB, CC) or −4 (Centers CA, CD) fluorimeters (Thermo Fisher Scientific, Waltham, MA, United States) (Supplementary Table S1).
Next-generation-sequencing (NGS) analyses
The routine pre-analyses were performed using Illumina or IonTorrent sequencing instruments together with different panels and analysis pipelines. All analyses used the human reference genome version 19 (hg19) for the alignment and annotation processes. The establishment/validation of the custom nNGMv2 panel analyses were conducted as follows. Briefly, the DNA quality control score (DNA QC Score) of each sample was analyzed using Archer PreSeq DNA QC KITs (ArcherDX) using 5 µL of 1 ng/μL DNA as input. A DNA QC score limit of a maximum of 13.5 was chosen. The recommended DNA input (9.4–359.0 ng; median 102.0 ng) for library preparation based on the QC score was calculated using the PreSeq Calculator.1 NGS library preparations were performed according to the Archer VariantPlex Somatic Protocol for Illumina. Library concentration and quality were measured employing Qubit3- (Centers CC, CB) or −4 fluorimeters (Centers CA, CC, CD) (Thermo Fisher Scientific) and 2100 Bioanalyzer (Center CB, CD) or Tapestation instruments (Center CA) (Agilent Technologies, Santa Clara, CA, United States). Subsequently, libraries were sequenced on different Illumina instruments using appropriate flow cells, different library loading concentrations, cluster densities and clusters passing filters. FastQ files were generated from raw sequencing data employing Local Run Manager off-instrument 2.0 (Centers CC, CD; Illumina) and compiled to a single FastQ file per read direction per case by using a self-written python-script (FASTQ Merger v.1.0, CC) or other applications. All further analyses, including quality control and variant calling, were performed applying the cloud-based instance Archer Analysis Unlimited Version 6.2.7. Amongst others, the following sequencing quality metrics were applied: 1) mean target coverage ≥ 100; 2) unique fragment filtered on target percent ≥ 80%; 3) unique fragment total ≥ 150,000; 4) average unique DNA start sites per GSP2 ≥ 50. Analyses that did not pass all four quality limits were evaluated individually by including additional parameters presented in Supplementary Table S2.
Variant calling, annotation and evaluation
For the initial routine analysis different analysis software and variant calling tools were used. The cut offs for minimum allele frequencies (AF) of previously reported variants were: Center CA 5%, CB 2% (1% at low tumor cell content), CC 3% and CD 5%.
During the validation of the custom nNGMv2 panel the Archer Analysis 6.2.7 default “somatic” filter applying a minimum AF of 2.7% was used. For the detection of MET intron variants potentially resulting in MET exon 14 skipping a custom filter was used with the following additional settings: Filter Consequence like “intron_variant”; Symbol is MET. No filter was used for variants reported in the initial routine analysis with AF below 2.7%.
Variant annotation was performed using the Archer Analysis 6.2.7 as well as the IGV (Integrative Genomics Viewer, Broad Institute, Boston, MA, United States [10]) and the COSMIC (catalogue of somatic mutations in cancer (Sanger-Cancer Center, Cambridge, United Kingdom [11]) database.
Clinical relevance of the identified tumor variants was classified by the standard evaluation algorithm of each center including the ClinVar [12], COSMIC [11] JAX CKB,2 OncoKB [13] and Varsome [14] databases/tools. Only likely pathogenic, pathogenic and VUS (variant of unknown significance) variants were reported. Gene variants being a predictive biomarker for EMA or FDA approved targeted therapies were considered “clinically relevant.” Detailed information on identified variants can be found in Supplementary Table S1.
Statistical analysis
For the correlation of sequencing parameters Spearman’s test was applied using Graphpad Prism 8 (GraphPad Software, Inc., San Diego, CA, United States). Rank correlation values were considered as: very strong relationship: >0.7, strong: 0.40–0.69, moderate: 0.30–0.39, weak: 0.20–0.29. p-values lower than 5% (p < 0.05, two-sided) were defined as statistically significant.
Results
Design and specifications of the custom ArcherDX VariantPlex nNGMv2 panel
The nNGM (national Network Genomic Medicine lung cancer) demands the detection of defined areas including variant hot spots in 26 Genes (ALK, BRAF, CTNNB1, EGFR, ERBB2, FGFR1, FGFR2, FGFR3, FGFR4, HRAS, IDH1, IDH2, KEAP1, KRAS, MAP2K1, MET, NRAS, NTRK1, NTRK2, NTRK3, PIK3CA, PTEN, RET, ROS1, STK11, TP53). Alterations in these genes are associated with targeted therapies either approved for NSCLC or other solid tumor entities (off-label options) by the EMA, being studied in clinical trials or having other clinical indications (TP53). Biomarkers and associated EMA approved drugs or targeted inhibitors are summarized in Table 1.
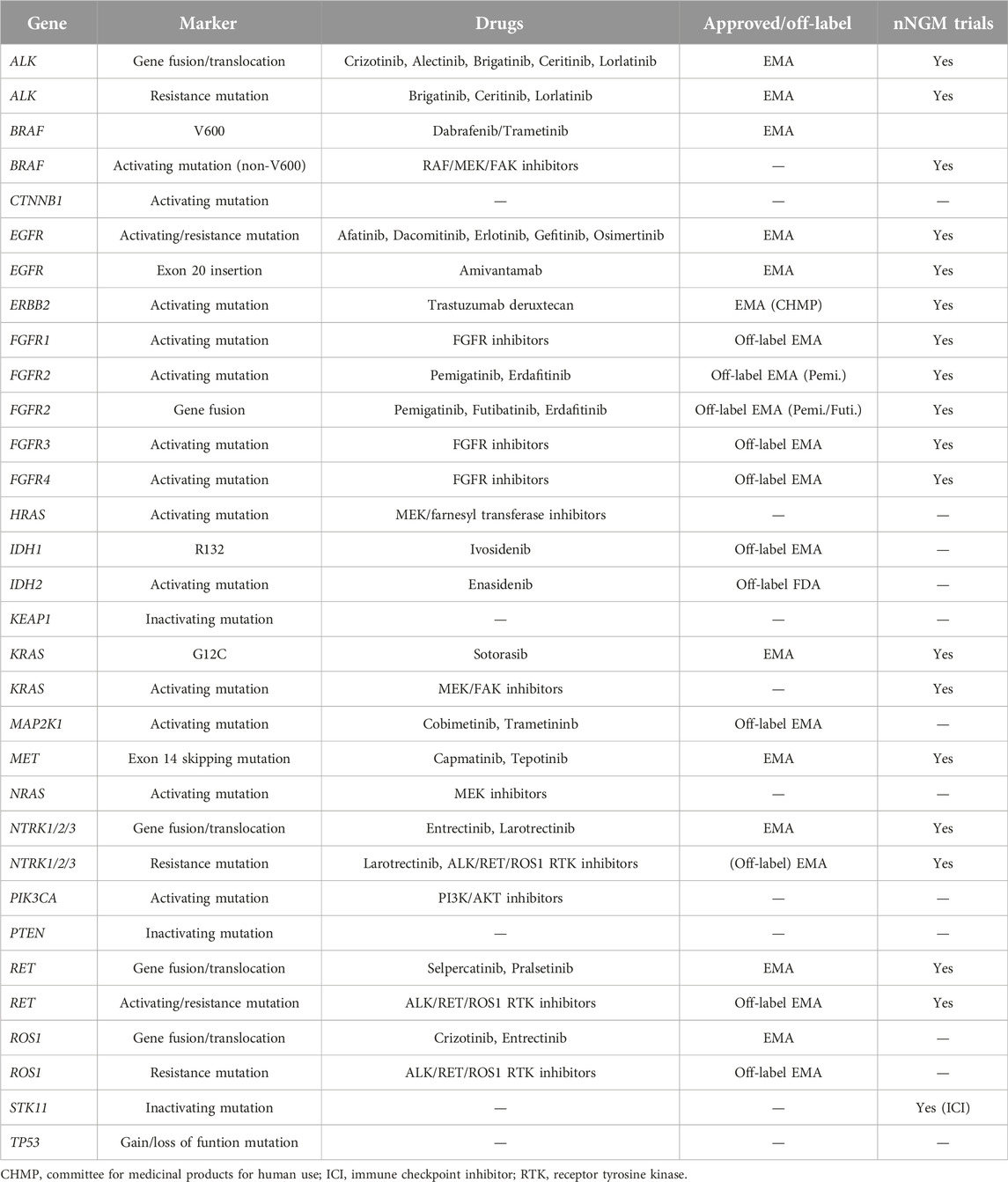
Table 1. Covered genes and associated EMA approved drugs or targeted inhibitors.
Notably, for the detection of variants resulting in exon 14 skipping of the MET gene coverage of the whole intron 13 and exon 14 as well as 100 bp of intron 14 of the MET gene are required to cover also non canonical splice site variants located more distant from the consensus splice sites. Moreover, the nNGMv2 panel covers 12 additional single nucleotide polymorphisms [SNP, rs17793354, rs987640, rs2269355, rs321198, rs338882, rs3780962, rs6444724, rs6811238, rs9951171, rs233214 (X-Chr), rs4829207 (X-Chr), Yp11.2 (Y-Chr)] for genetic identification and verification of tissue (ID-marking) especially in situations of multiple or resistance testing.
Therefore, a specific genomic content (exons, chromosomal regions) was defined on behalf of the nNGM (AH, Cologne) and subsequently converted into the custom nNGMv2 sequencing panel based on the Archer AMP technology by Archer’s design team. This panel includes unique molecular identifiers (UMIs) and was validated by the molecular pathology laboratories of the four nNGM centers University of Frankfurt (center CA), Giessen (CB), Munich (CC) and Ulm (CD). Detailed information on the covered genes, exons/introns and exact chromosomal areas are presented in Supplementary Table S3.
Study design and validation approach
The study design is depicted in Figure 1. First, each center assembled a list of NSCLCs which were pre-analyzed during routine diagnostics following nNGM regulations. A subset of cases (N = 93) was selected encompassing a large repertoire of tumor genetic variants relevant in NSCLC treatment and the majority of genes present in the nNGMv2 panel. Second, DNA quality was determined by qPCR (Archer PreSeq DNA QC, quality score had to be CT ≤ 13.5) allowing N = 90 (96.8%) to pass to the third step, the NGS of the samples using the newly designed nNGMv2 panel. MiniSeq (CB), MiSeq (CA, CB, CD) or NextSeq500/550 (CB, CC) sequencing devices (all Illumina) were used (Table 2). Fourth, for reasons of comparability, all results were analyzed applying the same Archer Analysis 6.2.7 software. N = 89 (98.9%) of the tested cases passed the Archer analysis pipeline and study internal quality requirements (Table 3, average values from all samples; Supplementary Table S2, values for each sample). Fifth, the resulting variant calls were annotated and evaluated for clinical relevance using not only Archer Suite’s built-in annotation algorithm, but also IGV, COSMIC, Clinvar, JAX CKB, OnkoKB and Varsome. Sixth, in a final step these results were compared to those of the original analyses (Figure 1). Finally, for the determination of sensitivity and robustness of the nNGMv2-panel 1) serial DNA dilution experiments of two cases (center CC) and 2) the Horizon OncoSpan FFPE standard (centers CB and CC) were included, respectively.
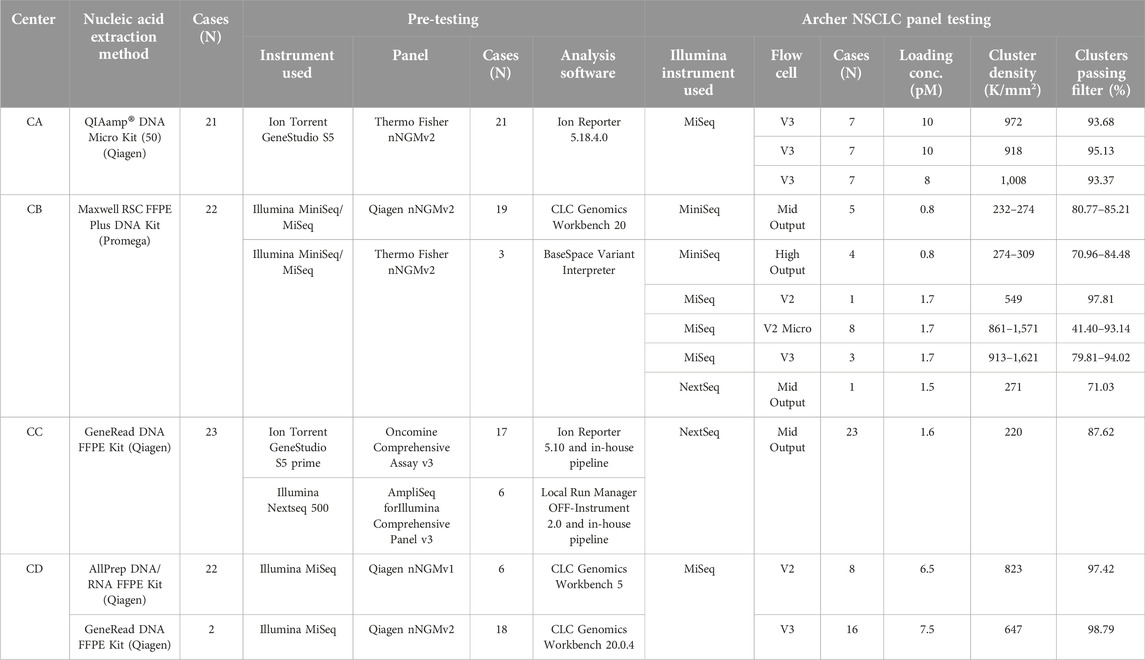
Table 2. Technical parameters.
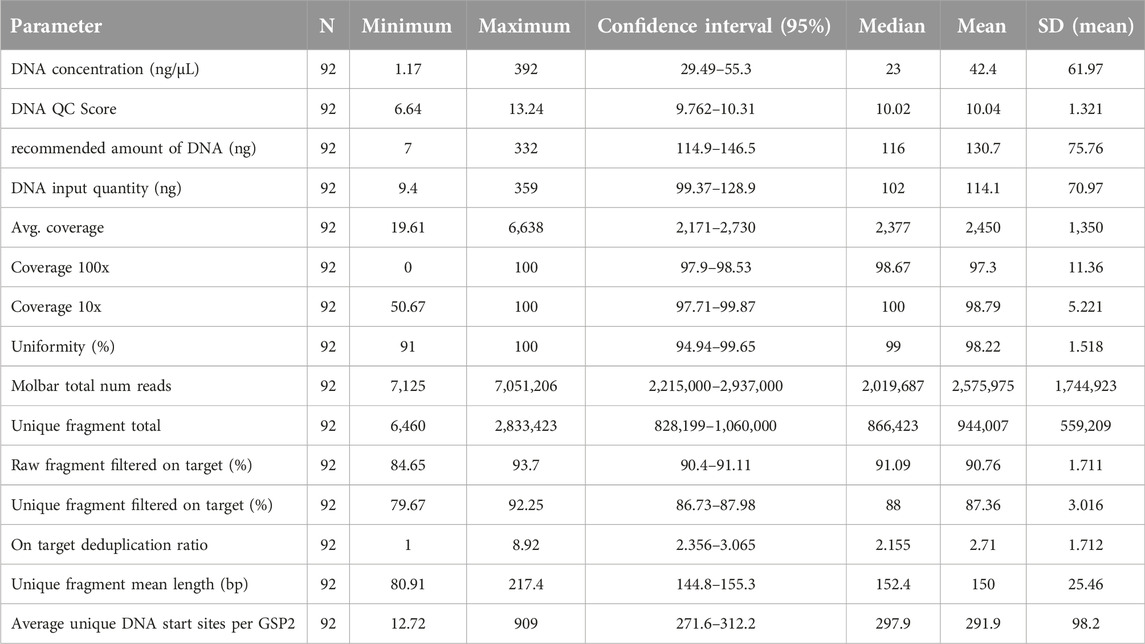
Table 3. Summary of quality control parameters of all samples.
Testing
All participating nNGM centers applied their own standard workflow during routine pre-testing and nNGMv2 analyses starting from nucleic acids extraction and ending with calling/annotation/evaluation of variants (Supplementary Table S1). Each center contributed more than 20 cases (21–26, average 22.5 per center, total N = 93, Figure 1 and Table 2) to this validation study. Subsequently, DNA quality was determined with Archer PreSeq DNA QC qPCR kits and the resulting DNA QC scores were transformed into DNA input as described in Materials and Methods. N = 90 (96.8%) DNA passed the test and were thus used as an input for NGS library preparation using nNGMv2 kits, sequencing, data processing and cloud-based Archer Analysis 6.2.7 as mentioned in Materials and Methods.
The Archer Analysis software 6.2.7 is available as an 1) on-premise installable virtual machine (VM, UNIX system) or 2) cloud-based instance (Archer Analysis Unlimited Version 6.2.7). In this study, a study-private instance of the cloud-based v6.2.7 was utilized for the analysis of each case. Resulting variants and annotations were revised according to the HGVS (Human Genome Variation Society [15]) nomenclature using IGV and COSMIC, which was especially necessary for variants affecting more than one base pair [multi-nucleotide variants (MNV), del, delins, dup, ins]. As mentioned above, each center evaluated the clinical/biological relevance based on their default classification system. In addition, variants, complying with HGVS rules, from all centers were submitted together with their reference NM-ID (MANE sequence) to ClinVar retrieving their pathogenic potential based on ClinVar evaluation criteria utilizing a NCBI Application Programming Interface (API)3 in an iterative manner applying a self-written python-code (PathInfony v.1.4.4, CC) (Supplementary Table S1).
Quality control (QC)
As DNA isolated from FFPE tissue is usually of lower quality compared to fresh tissue it might be important to know the usefulness of individual DNA isolates. Information about the isolated DNA can be obtained from different parameters including DNA concentration measurement and DNA QC scores (Archer PreSeq DNA QC). Parameters indicating the quality of NGS libraries are amongst others average coverage and mean length of unique fragments of the libraries (Table 3). Expectedly, the average number of unique start sites per GSP2 (gene specific primer) displayed a very strong correlation with average coverage (Spearman coefficient 0.815, p < 0.0001, Figure 2A) indicating the usefulness of these parameters given by the Archer analysis pipeline. Neither DNA concentration nor DNA QC score correlated with average coverage (Figures 2B, D) nor fragment length (Figures 2E, F). Only total amounts of DNA used as input for NGS library preparation showed a strong correlation with average coverage (Spearman coefficient 0.48, p < 0.0001, Figure 2C).
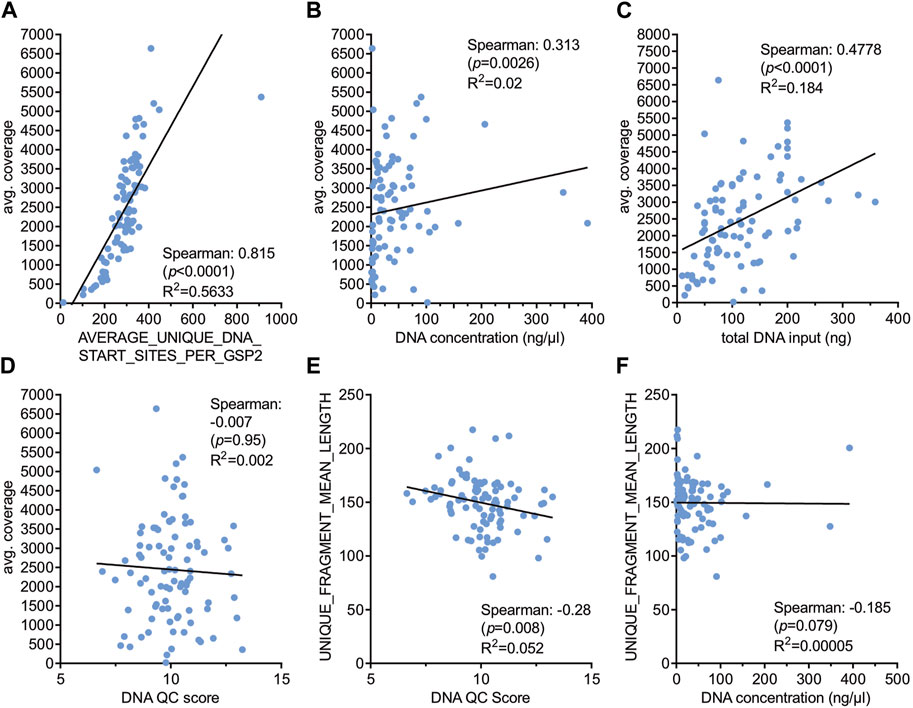
Figure 2. Correlation of various technical parameters with sequencing quality. Spearman correlation was performed with the indicated parameters (A–F) and correlation coefficients, p-values and R2 values are shown. DNA QC score was determined with the Archer PreSeq DNA QC KIT as described in Materials and Methods.
Characteristics of the VariantPlex nNGMv2 panel
To test whether reduced DNA concentration but similar total DNA input amounts influence the performance of the panel (Figure 3, Supplementary Table S4) a serial dilution of DNA from case CC-02-28, harboring a KRAS G12C alteration, was performed (44.0, 4.4, 2.6 and 0.9 ng/μL) and similar amounts of total DNA from each dilution were used for library preparation. No substantial changes regarding 1) average coverage (Figure 3A), 2) on target deduplication (Figure 3C) and 3) other parameters (Figure 3B) as well as 4) the KRAS G12C specific coverage and AF (Figure 3D) were observed indicating a high robustness of this panel. Next, a serial reduction of total input DNA (about 200, 100, 50, 25, 12.5 and 6.25 ng) of case CC-03-29 with an EGFR Exon 19 deletion was conducted (Figure 4, Supplementary Table S4). As expected, a continuous coverage reduction at lower DNA amounts was observed. However, even at 6.25 ng total input DNA an average UMI coverage of 494 (Figure 4A) and a target specific coverage for the EGFR Exon 19 deletion, much higher than the nNGM requirements for UMI panels (∼100), of 697 at similar AF (Figure 4D) were achieved. Expectedly, deduplication rates, an indirect measure for the multiplicity of genomes, increased notably below 25 ng DNA input (Figure 4C). Inversely corresponding, average unique DNA start sites per GSP2 were greatly reduced below 25 ng DNA to 134 at 6.25 ng but still much higher than the threshold of 50 (Figure 4B).
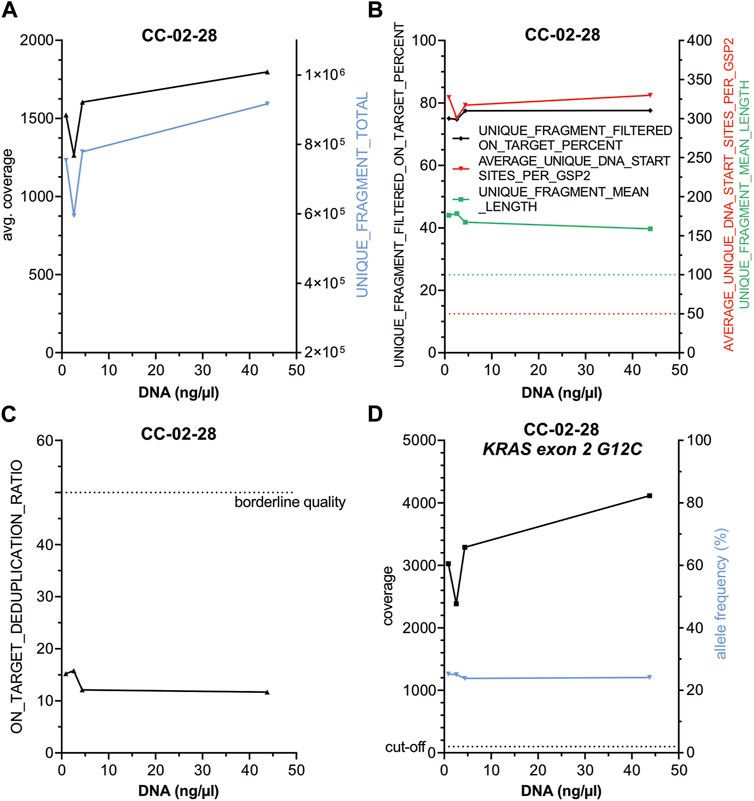
Figure 3. Reduction of DNA concentration does not influence the performance of the nNGMv2 panel. DNA from CC-02-28, harboring a KRAS G12C alteration, was diluted to 44, 4.4, 2.6 and 0.9 ng/μl and similar total amounts of DNA (44–50 ng) were used for library preparation. DNA concentration was plotted against the indicated sequencing quality parameters (A–D). Dashed lines, cut-offs of the shown parameters.
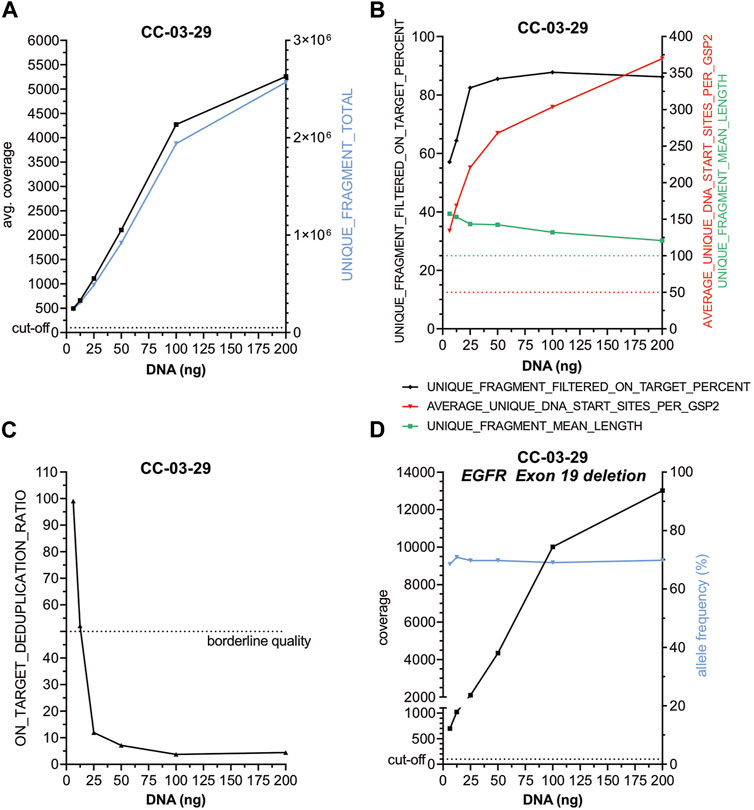
Figure 4. The AMP nNGMv2 panel delivers robust sequencing results with DNA input as low as 6.25 ng. Two hundred, 100, 50, 25, 12.5 and 6.25 ng DNA from case CC-03-29 harboring an EGFR Exon 19 deletion were used for library preparation. DNA amount was plotted against the indicated sequencing quality parameters (A–D). Dashed lines, cut-offs of the shown parameters.
Taken together, these data indicate that the nNGMv2 panel performance is excellent and stable covering a broad range of DNA input conditions starting with an input as low as an equivalent of 1,000 cells in a reliable manner even in situations where the input material is of lower quality.
Performance of the nNGMv2 Archer analysis pipeline—testing of a pre-defined FFPE control tissue
In order to evaluate accuracy and specificity of the nNGMv2 panel two centers (CB and CC) subjected the Horizon OncoSpan FFPE control, comprising 13 variants that were cross validated by Horizon with NGS and digital droplet PCR and covered by this panel, to NGS (Supplementary Table S5). All 13 alterations, including three deletions, were identified by both centers and the majority of the variants (80.8%) were detected at AF within the acceptance criteria. Only one variant (PIK3CA E545K) was called by both centers at AF outside the acceptance range (7.00%–10.60%; CB, −3.71%; CC, +18.01%).
Performance of the nNGMv2 Archer analysis pipeline—comparison with pre-testing results
To investigate the performance, including specificity (variant calling) and precision (AF), of the nNGMv2 panel, the results of the Archer Analysis pipeline were compared with the outcomes of the previous routine diagnostics (N = 89; Table 4). QC parameters and variant calling limits as well as the variant evaluation algorithm are described in detail in Materials and Methods. The pre-testing of the cases included different analysis pipelines including IonReporter, Qiagen CLC Workbench and an in-house Illumina pipeline (Tables 2, 4). Sequencing quality of the nNGMv2 panel analyses and of the various utilized Illumina systems are presented in Table 2 per center. The analyzed samples displayed tumor cell contents ranging from 2% to 90% (median = 50%) (Supplementary Table S1). In total, 192 variants were called in the pre-testing analyses in almost all (24—ALK and HRAS missing) of the 26 genes present in the nNGMv2 panel including 25 deletions (EGFR, KEAP1, MET, PIK3CA, PTEN, STK11, TP53), nine deletion insertions (EGFR, FGFR2, IDH1, KRAS, MET, TP53), six duplications (EGFR, KEAP1, STK11, ERBB2) and seven splice site variants (MET, TP53). All alterations but two MET intron 13 non-consensus splice variants that might be leading to exon 14 skipping (not confirmed on RNA level) were identified with the Archer Analysis default filter settings. When applying an adapted filter for MET intron variants (see Methods) both variants were called. Moreover, three additional variants that were in principle covered by the employed NGS panel (KEAP1 and STK11 truncation variants and a TP53 point mutation) but not called during pre-testing by the utilized algorithms were identified with the nNGMv2 panel. An excellent precision of the developed panel regarding AF of the variants was also observed. The median AF difference was 0.29% and only 13 variants displayed a difference in AF greater than 10%. In summary of all validation steps, the nNGMv2 panel displayed an excellent specificity, precision as well as robustness. The panel was successfully applied to samples with low cell numbers/DNA content. However, an adapted/second filter was required for the detection of certain more distant non-canonic MET exon 14 splice site alterations [16].
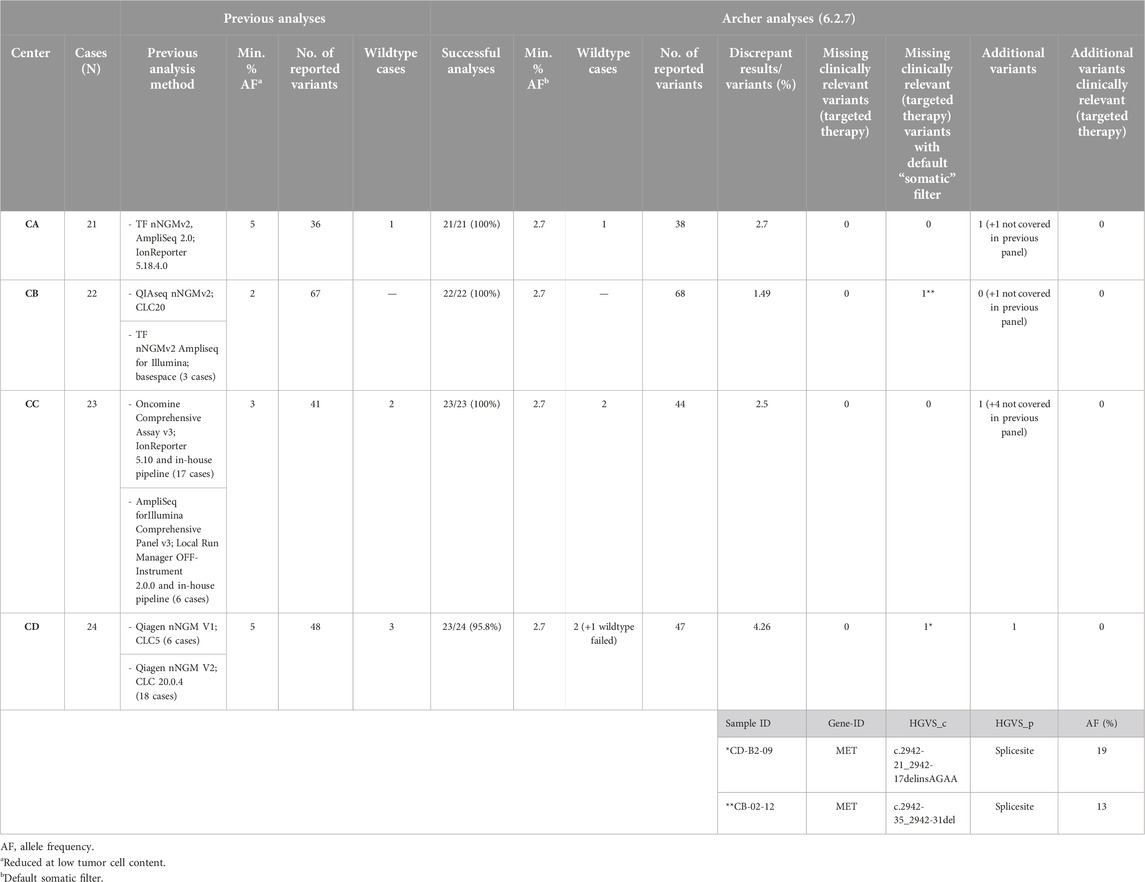
Table 4. Summary: comparison with previous results.
Discussion
Here, the AMP-based nNGMv2 panel was established and validated in a multicentric approach for potential diagnostic testing of NSCLC patient samples fulfilling the nNGM analysis requirements [7].
The AMP technique applies sample and molecular (unique molecular identifier) barcode adapter ligation to the DNA followed by usually two subsequent semi-nested PCRs each employing a different gene-specific primer (GSP1, GSP2) in combination with universal primers [9]. AMP offers several advantages compared to AMPL and HCP: 1) amplification is highly specific due to a semi-nested approach with two GSPs reducing off-target amplification, 2) easy multiplexing significantly increasing throughput and cost-effectiveness of NGS library preparation. 3) works with reduced input requirements (both amounts and quality) which is particularly beneficial when working with limited FFPE NSCLC biopsies routinely employed for molecular pathology analyses. 4) high flexibility in target selection making it easy to adapt existing panels, 5) high uniformity of libraries, 6) simple hands-on protocol. Therefore, AMP 7) is easily scalable, 8) has a fast turnaround time as summarized by Zheng et al. [9].
Here, we compare results obtained with the AMP approach (nNGMv2 panel) to results from previous analyses performed during routine testing with AMPL techniques. A direct head-to-head comparison of the techniques was not possible due to lack of sufficient DNA amounts. Nevertheless, both the pre and the AMP nNGMv2 panel testing were performed with the same DNA. Initially, 24–30 samples were selected out of more than 200 recently pre-tested (AMPL) DNAs per center. The samples were selected to encompass various different tumor genetic variants, including complex insertions/deletions and splice site variants, as well as a broad range of DNA concentrations/amounts and tumor cell contents. Moreover, sensitivity (DNA input/cell number) and precision (allele frequency) of the panel were determined by library preparation from serial dilutions of DNA from two cases or a Horizon standard, respectively. We confirmed that the AMP-based nNGMv2 panel in combination with the Archer analysis suite provides specific, reliable and robust results at high uniformity of the sequencing results even with small amounts of input material (DNA input as low as 6.25 ng/∼1,000 cells).
Sequencing was successful in 98.9% of the samples (N = 90) even though small biopsies resulting in low DNA concentrations of <5 ng/μL (mean 42.4 ng/μL) in 21% of the samples were included. The sequencing analysis of one sample at a high DNA concentration (102 ng/μL) failed probably due to technical problems. Since all samples were previously tested at least at acceptable quality values and thus preselected, a comparison of both techniques (AMP vs. AMPL) regarding performance cannot be performed. However, after completion of the validation study, Center CC used the AMP-based nNGMv2 panel for routine testing of about 1,000 NSCLC cases until the end of 2022. Initially, extracted DNAs from all FFPE samples were used independent of DNA concentrations for library preparation and subsequent sequencing. Nevertheless, after re-evaluation of the sequencing qualities of the first 100 cases the minimum DNA concentration threshold was set to 2 ng/μL for subsequent cases achieving >96% successful analyses (data not shown). This success rate is high in comparison with the experiences at center CC with various Ion Torrent Oncomine Assays where only 85%–90% of the sequencing analyses were successful at the same DNA concentration cutoff (data not shown). Moreover, it is possible to mix AMP NGS libraries prepared with different techniques, e.g., nNGMv2 or other panels of this type (AMP), Ampliseq for Illumina BRCA- (AMPL) as well as TSO500 HRD-panels (HCP) on a single flow cell in parallel thereby increasing flexibility. Nevertheless, it is recommendable to develop a system specific (Illumina device, flow cell, number of samples) dilution protocol before pooling all libraries as undiluted Archer AMP libraries may lead to irregular clustering depending on the amount of samples.
All but two potential MET exon 14 skipping variants were identified at similar allele frequencies with default filter settings in the Archer analysis suite. Both MET variants were found with an adapted calling filter showing that, as experienced with analysis pipelines from other sequencing systems, adjustment/refinement of the analysis algorithm is also required to obtain correct/complete results. Three additional variants (KEAP1, STK11, TP53) were called with the nNGMv2 panel that were not identified in the pre-testing analyses. For example, the STK11 truncation variant was not called by the Ion Reporter (5.10) although clearly present in IGV (coverage 1,463, AF 18.9%). Although all three variants do not represent direct targets for approved targeted therapies their detection is important because they might influence the efficacy of immune checkpoint inhibitors in the context of other mutations [17–19]. In this context, our results showed a similar if not even slightly better performance of the Archer analysis suite compared with analysis systems utilized in pre-testing.
It is still an often-discussed question if and how to control quality of input material for generation of NGS-libraries. Several different approaches are available including qPCR measurements of various genes, DNA bio-analyzer profile, DNA ladder amplification and/or DNA quantification [20]. In our study, we demonstrated that using total amounts of DNA as quality determining parameter is superior to a real time PCR based approach (here: Archer PreSeq DNA QC KIT). If this situation is different when applying other qPCR kits or approaches has to be tested experimentally.
Our validation study has clear limitations which are majorly due to the lack of tissue and sufficient DNA amounts as well as the high cost of NGS analyses. 1) For these reasons, a direct head-to-head comparison of the techniques (pre and AMP test) was not feasible, an inter-laboratory exchange (ring trial) was not performed and only limited intra or inter reproducibility tests were included [21]. However, the results obtained with our study design clearly showed the excellent performance of the VariantPlex nNGMv2 panel. Moreover, center CC successfully completed various external ring trials hosted by the Quality in Pathology (QuIP, Berlin, Germany) and the required internal nNGM performance and proficiency tests (including the multigene ring trial) applying the VariantPlex nNGMv2 panel. 2) The VariantPlex system does not cover the detection of gene fusions/translocations and thus is insufficient as a stand-alone biomarker test for NSCLC. Therefore, the nNGM algorithm includes parallel analysis systems for gene fusion detection such as ArcherDx FusionPlex NGS tests or FISH. Recently, ArcherDx expanded the FusionPlex system to perform parallel testing for small nucleotide variants and fusions on RNA level [22]. However, in our hands the coverage of key driver mutations is not optimal, and the corresponding allele frequencies differ significantly from those obtained on DNA level (data not shown). Since the FusionPlex Lung v2 panel is performed in parallel to DNA analysis for detection of relevant gene fusions (ALK, MET, NTRK1/2/2, RET, ROS1), it is rather recommendable for confirmation of variants identified with the VariantPlex analysis.
Taken together, we show that the AMP technology is not only versatile and easy to be used in daily routine diagnostics of NGS-panel based detection of actionable variants in lung cancer but is also a robust technique which can handle DNA of different sources and a broad range of qualities. Thus, AMP represents a suitable workhorse in the daily routine care of molecular pathology.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by the ethics committees of the faculties of medicine of the LMU Munich (project number: 19-542) and the Goethe University Frankfurt (project number: 19-425). The patients provided written informed consent to participate in this study. The study was conducted in accordance with the Declaration of Helsinki.
Author contributions
JK: Conceptualization, data curation, formal analysis, investigation, methodology, project administration, supervision, writing—original draft. M-CD: Conceptualization, data curation, formal analysis, investigation, methodology, writing—review editing. JJ: data curation, formal analysis, investigation. AB: Conceptualization, data curation, formal analysis, investigation, methodology, writing—review editing. KH: data curation, formal analysis, investigation. UG: Conceptualization, data curation, formal analysis, investigation, methodology, writing—review editing. RM: Conceptualization, investigation, writing—review editing. AH: formal analysis, investigation, panel design. NB, CL, and FF: Conceptualization, investigation, methodology, resources, writing—review editing. PW, SG, PM, and FK: Conceptualization, resources, writing—review editing. AJ: Conceptualization, investigation, methodology, project administration, supervision, writing—original draft.
Funding
Archer VariantPlex nNGMv2 sequencing kits, Archer PreSeq DNA QC kits and Horizon OncoSpan controls were provided by ArcherDx.
Conflict of interest
AJ received honoraria for scientific talks, participance in adboards and reimbursement of travel as well as accommodation expenses from Amgen, AstraZeneca, Bayer Pharmaceuticals, BMS, Biocartis, Boehringer Ingelheim, Merck KgA, Lilly, MSD, Novartis, QuIP GmbH, Roche Pharma, Stemline, Takeda. M-CD has received consulting fees and honoraria for lectures by Biocartis, Roche, Bayer, Janssen-Cilag, Novartis, Thermo Fisher Scientific, Molecular Health, Qiagen and Astra Zeneca. Research Support was provided by Astra Zeneca, Roche and Thermo Fisher. RM received a research grant from BMS and participated in adboards from BMS and AstraZeneca. CL, FF, and NB are affiliated with ArcherDx which supported the study.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author disclaimer
The authors are fully responsible for the content of this paper, and the views and opinions described in the publication solely reflect those of the authors.
Acknowledgments
We thank Konstanze Schleich for technical and organizational support.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.por-journal.com/articles/10.3389/pore.2024.1611590/full#supplementary-material
Footnotes
1https://eu.idtdna.com/pages/support/archer-support
3https://www.ncbi.nlm.nih.gov/home/develop/api/
References
1. Falcone, R, Lombardi, P, Filetti, M, Duranti, S, Pietragalla, A, Fabi, A, et al. Oncologic drugs approval in europe for solid tumors: overview of the last 6 years. Cancers (Basel) (2022) 14:889. doi:10.3390/cancers14040889
2. Oda, Y, and Narukawa, M. Response rate of anticancer drugs approved by the Food and Drug Administration based on a single-arm trial. BMC Cancer (2022) 22:277. doi:10.1186/s12885-022-09383-w
3. Turner, JH. An introduction to the clinical practice of theranostics in oncology. Br J Radiol (2018) 91:20180440. doi:10.1259/bjr.20180440
4. Herrera-Juarez, M, Serrano-Gomez, C, Bote-de-Cabo, H, and Paz-Ares, L. Targeted therapy for lung cancer: beyond EGFR and ALK. Cancer (2023) 129:1803–20. doi:10.1002/cncr.34757
5. Khadela, A, Postwala, H, Rana, D, Dave, H, Ranch, K, and Boddu, SHS. A review of recent advances in the novel therapeutic targets and immunotherapy for lung cancer. Immunotherapy Lung Cancer Med Oncol (2023) 40:152. doi:10.1007/s12032-023-02005-w
6. Morash, M, Mitchell, H, Beltran, H, Elemento, O, and Pathak, J. The role of next-generation sequencing in precision medicine: a review of outcomes in oncology. J Pers Med (2018) 8:30. doi:10.3390/jpm8030030
7. Buttner, R, Wolf, J, Kron, A, and Nationales Netzwerk Genomische, M. The national Network Genomic Medicine (nNGM): model for innovative diagnostics and therapy of lung cancer within a public healthcare system. Pathologe (2019) 40:276–80. doi:10.1007/s00292-019-0605-4
8. Singh, RR. Target enrichment approaches for next-generation sequencing applications in oncology. Diagnostics (Basel) (2022) 12:1539. doi:10.3390/diagnostics12071539
9. Zheng, Z, Liebers, M, Zhelyazkova, B, Cao, Y, Panditi, D, Lynch, KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med (2014) 20:1479–84. doi:10.1038/nm.3729
10. Robinson, JT, Thorvaldsdottir, H, Wenger, AM, Zehir, A, and Mesirov, JP. Variant review with the integrative genomics viewer. Cancer Res (2017) 77:e31–4. doi:10.1158/0008-5472.CAN-17-0337
11. Tate, JG, Bamford, S, Jubb, HC, Sondka, Z, Beare, DM, Bindal, N, et al. COSMIC: the catalogue of somatic mutations. Cancer Nucleic Acids Res (2019) 47:D941–D947. doi:10.1093/nar/gky1015
12. Landrum, MJ, Lee, JM, Benson, M, Brown, GR, Chao, C, Chitipiralla, S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res (2018) 46:D1062–D1067. doi:10.1093/nar/gkx1153
13. Chakravarty, D, Gao, J, Phillips, SM, Kundra, R, Zhang, H, Wang, J, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol (2017) 2017:1–16. doi:10.1200/PO.17.00011
14. Kopanos, C, Tsiolkas, V, Kouris, A, Chapple, CE, Albarca Aguilera, M, Meyer, R, et al. VarSome: the human genomic variant search engine. Bioinformatics (2019) 35:1978–80. doi:10.1093/bioinformatics/bty897
15. den Dunnen, JT, Dalgleish, R, Maglott, DR, Hart, RK, Greenblatt, MS, McGowan-Jordan, J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat (2016) 37:564–9. doi:10.1002/humu.22981
16. Awad, MM, Oxnard, GR, Jackman, DM, Savukoski, DO, Hall, D, Shivdasani, P, et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-met overexpression. J Clin Oncol (2016) 34:721–30. doi:10.1200/JCO.2015.63.4600
17. West, HJ, McCleland, M, Cappuzzo, F, Reck, M, Mok, TS, Jotte, RM, et al. Clinical efficacy of atezolizumab plus bevacizumab and chemotherapy in KRAS-mutated non-small cell lung cancer with STK11, KEAP1, or TP53 comutations: subgroup results from the phase III IMpower150 trial. J Immunother Cancer (2022) 10:e003027. doi:10.1136/jitc-2021-003027
18. Marinelli, D, Mazzotta, M, Scalera, S, Terrenato, I, Sperati, F, D'Ambrosio, L, et al. KEAP1-driven co-mutations in lung adenocarcinoma unresponsive to immunotherapy despite high tumor mutational burden. Ann Oncol (2020) 31:1746–54. doi:10.1016/j.annonc.2020.08.2105
19. Ricciuti, B, Arbour, KC, Lin, JJ, Vajdi, A, Vokes, N, Hong, L, et al. Diminished efficacy of programmed death-(ligand)1 inhibition in STK11- and KEAP1-mutant lung adenocarcinoma is affected by KRAS mutation status. J Thorac Oncol (2022) 17:399–410. doi:10.1016/j.jtho.2021.10.013
20. Dang, J, Mendez, P, Lee, S, Kim, JW, Yoon, JH, Kim, TW, et al. Development of a robust DNA quality and quantity assessment qPCR assay for targeted next-generation sequencing library preparation. Int J Oncol (2016) 49:1755–65. doi:10.3892/ijo.2016.3654
21. Jennings, LJ, Arcila, ME, Corless, C, Kamel-Reid, S, Lubin, IM, Pfeifer, J, et al. Guidelines for validation of next-generation sequencing-based oncology panels: a joint consensus recommendation of the association for molecular pathology and college of American pathologists. J Mol Diagn (2017) 19:341–65. doi:10.1016/j.jmoldx.2017.01.011
22. Desmeules, P, Boudreau, DK, Bastien, N, Boulanger, MC, Bosse, Y, Joubert, P, et al. Performance of an RNA-based next-generation sequencing assay for combined detection of clinically actionable fusions and hotspot mutations in NSCLC. JTO Clin Res Rep (2022) 3:100276. doi:10.1016/j.jtocrr.2022.100276
Keywords: lung cancer, targeted therapy, next-generation sequencing panel, anchored multiplex PCR, actionable mutations
Citation: Kumbrink J, Demes M-C, Jeroch J, Bräuninger A, Hartung K, Gerstenmaier U, Marienfeld R, Hillmer A, Bohn N, Lehning C, Ferch F, Wild P, Gattenlöhner S, Möller P, Klauschen F and Jung A (2024) Development, testing and validation of a targeted NGS-panel for the detection of actionable mutations in lung cancer (NSCLC) using anchored multiplex PCR technology in a multicentric setting. Pathol. Oncol. Res. 30:1611590. doi: 10.3389/pore.2024.1611590
Received: 16 November 2023; Accepted: 15 March 2024;
Published: 28 March 2024.
Edited by:
József Tímár, Semmelweis University, HungaryCopyright © 2024 Kumbrink, Demes, Jeroch, Bräuninger, Hartung, Gerstenmaier, Marienfeld, Hillmer, Bohn, Lehning, Ferch, Wild, Gattenlöhner, Möller, Klauschen and Jung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jörg Kumbrink, am9lcmcua3VtYnJpbmtAbWVkLnVuaS1tdWVuY2hlbi5kZQ==