 Muhammad Ihtisham Umar1Waseem Hassan1Ghulam Murtaza1*Manal Buabeid2,3*Elshaimaa Arafa2,3Hafiz Muhammad Irfan4Mohd Zaini Asmawi5Xianju Huang6
Muhammad Ihtisham Umar1Waseem Hassan1Ghulam Murtaza1*Manal Buabeid2,3*Elshaimaa Arafa2,3Hafiz Muhammad Irfan4Mohd Zaini Asmawi5Xianju Huang6- 1Department of Pharmacy, COMSATS University Islamabad, Lahore Campus, Lahore, Pakistan
- 2Department of Clinical Sciences, Ajman University, Ajman, United Arab Emirates
- 3Medical and Bio-allied Health Sciences Research Centre, Ajman University, Ajman, United Arab Emirates
- 4Department of Pharmacy, University of Sargodha, Sargodha, Pakistan
- 5School of Pharmaceutical Sciences, University of Science Malaysia, Pulau Pinang, Malaysia
- 6College of Pharmacy, South-Central University for Nationalities, Wuhan, China
A hormonal imbalance may disrupt the rigorously monitored cellular microenvironment by hampering the natural homeostatic mechanisms. The most common example of such hormonal glitch could be seen in obesity where the uprise in adipokine levels is in virtue of the expanding bulk of adipose tissue. Such aberrant endocrine signaling disrupts the regulation of cellular fate, rendering the cells to live in a tumor supportive microenvironment. Previously, it was believed that the adipokines support cancer proliferation and metastasis with no direct involvement in neoplastic transformations and tumorigenesis. However, the recent studies have reported discrete mechanisms that establish the direct involvement of adipokine signaling in tumorigenesis. Moreover, the individual adipokine profile of the patients has never been considered in the prognosis and staging of the disease. Hence, the present manuscript has focused on the reported extensive mechanisms that culminate the basis of poor prognosis and diminished survival rate in obese cancer patients.
Introduction
A sluggish lifestyle is often associated with a disequilibrium between calorie-intake and energy expenditure. Such disproportion accounts for an increase in the bulk of adipose tissue, principally the white adipose tissue (WAT) that eventually precipitates in obesity. WAT serves as a major fat reservoir wherein the upregulation of PPARγ and C/EBPα abet a rapid proliferation and differentiation of precursor cells into unilocular adipocytes [1]. More than 98% mass of these adipocytes is covered by a single lipid droplet, leaving less than 2% space for nucleus and organelles. Committed to store triglycerides in their fat droplets, these cells contain very few mitochondria and remain sedentary. Contrarily, the multilocular adipocytes of the brown adipose tissue (BAT) contain several smaller fat droplets and plenty of mitochondria in their cytoplasm. These adipocytes upregulate uncoupling protein-1 (UCP-1) that instigate heat generation via ‘non-shivering thermogenesis’ [2]. Previously, BAT was believed to exist in neonates exclusively, it is only recently that the anatomical location of BAT is reported in the axillary, perivascular, perirenal, cervical and supraclavicular regions in adults [3]. Under certain conditions such as hypothermia or exercise, some adipocytes in WAT are transformed into a third type of adipocytic lineage that is termed as “beige-like adipocyte”. Congeneric with BAT, the beige-like adipocytes also generate heat, yet they differ from BAT adipocytes in their location, lineage [4] and markers [5]. Aforesaid transformation of WAT into beige lineage is provoked by sympathetic stimulation via upregulation of bone morphogenetic protein 7 (BMP7) [6] and repression of Foxp1 through fibroblast growth factor signaling [7].
The unilocular adipocytes of WAT not only store fats, but they also release hormones that are termed as adipokines. Accordingly, an increasing bulk of WAT in obesity corresponds to a remarkable incline in adipokine levels. In the first place, the consistently higher glucose levels in obesity induce hyperinsulinemia. Secondly, the sensitivity of insulin receptors is dwindled down because of the upsurge in adipokine levels. Such decline in insulin sensitivity not only culminates in type 2 diabetes mellitus, the hyperinsulinemia and insulin resistance directly increase the risk of many cancers such as breast, endometrial, colorectal and pancreatic carcinoma. Consequently, the obese population is considered at higher risk for cancers. This ‘under-rated’ notion requires enormous public attention by virtue of the rapidly increasing obese population that has gone triple since 1975 as per the fact sheets of The World Health Organization [8]. In a cohort study on 1.2 million obese women with high body mass index (age 50–64 years), the occurrence of cancers such as breast, endometrial, colorectal, non-Hodgkin lymphoma, leukemia, ovarian and pancreatic carcinoma was found as much higher as 58.8% [9]. According to the United States population data of 2012, around 3.5% (28,000) of all annually reported cancers in men were associated with obesity [10]. The women were found even more vulnerable with around 9% (72,000) obesity derived cancers reported per year. In a parallel study, around 21% of all cancer cases reported in Nigeria between 2012 and 2014 were linked with obesity [11]. Although of relatively lower magnitude, the risk of pancreatic cancer in obese population was significantly higher than the non-obese subjects [12].
The regulation of cell cycle and its check points are presented in Figure 1. Under normal conditions, the cells remain in a reversible stationary phase (quiescence) until they are provided with optimum growing space, nutrients and growth factors [13]. Quiescence is associated with an upregulation of serum deprived early response genes (SDERGs) including tumor repressor gene SALL2 and MXI1 [14]. Once that the required factors are provided, the cells upregulate proliferative genes such as c-Myc and cylins that promote the entry of these previously quiescent cell to the cell cycle. The basic difference between the normal and cancer cells lies in the fact that the later become independent of either of the above-mentioned factors. Such lack of sensitivity to the mitogen availability and space makes the cancer cells grow beyond the limits, provoking drastic physiological modifications by inducing aberrant angiogenesis to support their freaky growth. Hence, the first deviation of the cancer cells from normal lineage is their escape from quiescence in mitogen deprived conditions. Despite of the detailed studies on the expression of different proliferative genes involved in the escape of cells from quiescence, the role of adipokine upswing in the dysregulation of quiescence check points necessitates immense consideration.
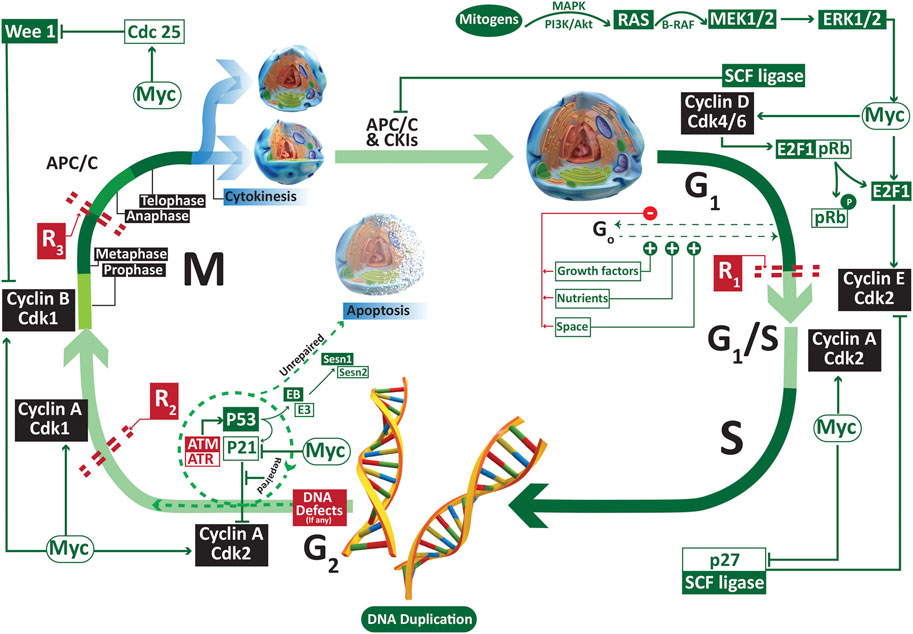
FIGURE 1. The regulators of G1, S, G2 and M phase of cell cycle. A normal cell remains dependent on external factors such as the nutrients, growth factors and growing space only in the early G1 phase. A lack of even one of these three factors may result into the reversible escape (Go or quiescence) of the cell from cell cycle. Once the cell crosses the first restriction phase R1, it becomes independent of the availability of the external factors. Abbreviations: APC/C anaphase promoting complex/cyclosome; ATM ataxia telangiectasia mutated; ATR ATM-and Rad3 related; Cdk cyclin dependent kinase; R restriction point.
In addition to the dysregulation of cell cycle regulation, the metastasis is another major impediment in the management of cancers. As an estimate, more than 90% of cancer caused deaths involve the spread of cancer cells from their site of origin to the distant locations [15]. The tendency to spread depends on many factors, one of which is the type of the disease itself. For instance, brain tumors and skin carcinomas seldom metastasize [16], albeit their high tendency to invade locally. On the other side, bone sarcomas metastasize quickly to other tissue, especially the lungs [17]. The different stages of metastasis are graphically presented in Figure 2. In general, the process of metastasis can be divided into four major stages, the local invasion, intravasation, spread and extravasation where local invasion is the primary step that paves way to the distant colonization of the cancer cells. Despite of being fully differentiated, the epithelial cancer cells have to endure a morphological shift from epithelial to mesenchymal attributes to invade the surrounding tissue through a process that is termed as the epithelial to mesenchymal transition (EMT). Although such EMT is a much-needed physiological phenomena in embryogenesis and wound healing, it sets the stage for the more detrimental distant translocation of cancer cells through blood or lymph vessels via intravasation. Once metastasized, these cells may undergo a reverse mesenchymal to epithelial transition (MET) that facilitates the successful colonization of these cells into a new distant site through extravasation. Since the role of some of the potential adipokines in instigating EMT and cell cycle dysregulation is established, we need to link all possible points on paper to fully understand the role of adipokine upregulation in the pathological quintessence of tumor microenvironment.
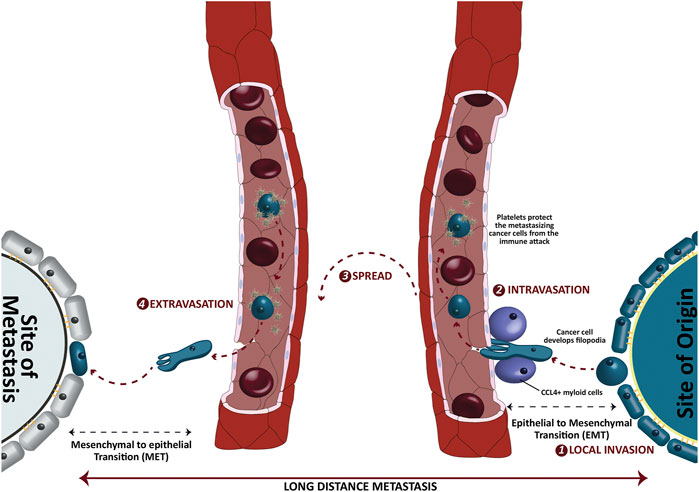
FIGURE 2. Different stages of long-distance metastasis of epithelial cancer cells. The cancer cells invade the surrounding tissues and intravasate a nearby blood capillary via chemokine ligand 4 positive (CCL4+) myeloid cells. Once entered in a capillary, the platelets protect these spreading cancer cells from a possible immune attack. The epithelial to mesenchymal transition (EMT) helps these cancer cells to downregulate the epithelial markers such as E-cadherins and develop filopodia. On the other side, the mesenchymal to epithelial transition (MET) upregulates the epithelial characters to get localized at the distant new location.
It is recently reported that the increasing bulk of fat in obesity is not the independent contributor to cancer pathogenesis. A cohort study on 319,397 Korean subjects has suggested that the people with obesity derived metabolic derangements are at relatively higher risk of colorectal cancer [18]. Despite of the high body mass index, the risk of colorectal cancers and cardiovascular ailments was remarkably minimum in a sub-population of obese subjects who did not show metabolic dysregulations such as high blood pressure, hyperinsulinemia, dyslipidemia, and insulin resistance. Such findings suggest a possible link between the metabolic dysregulation and the neoplastic shift among different tissues particularly the colorectal cells in both male and female subjects. It seems as if the the higher levels of adipokines, pro-inflammatory cytokines and hyperinsulinemia together shift the cellular microenvironment to an immune-suppressant tumor supportive environment that favors the survival, proliferation, and metastasis of cancer cells. Currently, the prognosis and staging of the cancer patients is made according to the tumor node metastasis (TNM) classification that is advised by the Union of International Cancer Control (UICC) [19]. The TNM classification provides current guidelines to classify the metastatic extent, however many inclusions have been suggested over the years to further improve disease prognosis. These suggested inclusions comprise of the genetic factors [20], deep proteomics [21], immunohistochemistry [22] and the molecular signatures [23]. Despite of the established role of obesity in cancer pathogenesis, the consideration of adipokine profile has never been suggested in the staging and profiling of the disease. In the next sections of the manuscript, we now focus on the different molecular mechanisms reported so far to establish the pathological basis of adipokines and obesity mediators in cancer.
Obesity-Derived Mediators and Cancer
The adipokines released from the adipocytes help these cells to communicate with surrounding tissues to accomplish the regulation of glucose and fat metabolism. Some of these adipokines are exclusively released from adipose such as leptin, resistin, visfatin and omentin whereas the others are secreted from other tissues as well. For instance adiponectin is released from placenta, myocytes, liver parenchymal cells and the osteoblasts. Likewise, tumor necrosis factor (TNF) and interleukin 6 (IL-6) are released from adipocytes and the infiltrating macrophages. Under normal physiological conditions, these adipokines perform valuable functions to maintain homeostasis. However, despite of their potential biological significance, there is a compelling scientific evidence that supports the involvement of these adipokines in the growth and spread of cancer cells. For instance, leptin is not expressed in the normal breast tissue. However, a growing ductal carcinoma induces a significant expression of leptin as a major proliferative factor in the surrounding normal breast tissue, suggesting a possible role of leptin in disease progression [24]. The higher expression levels of resistin and visfatin are reported in a number of malignancies such as colorectal [25], lung [26], gastroesophageal [27], endometrial [28] and breast cancer [29].
Apart from the above-mentioned mediators, there are other mediators whose serum level is often associated with obesity. These include angiopoietin (Ang) and angiopoieting like proteins (ANGLP), chemerins, endotropin, fibroblast growth factor 2, lipocalin 2, neuregulin 4, retinol binding protein 4, autotaxin and its derivative lysophosphatidic acid, bone morphogenic proteins, sphingolipids, fatty acid esters of hydroxyl fatty acids, uric acid and uridine [30]. The over-expression of these mediators is reported in several different malignancies (Please see Supplementary Material Table 1). Interestingly, some of these mediators are found to be specific for a particular type of cancer. For instance, leptin over-expression is found to be relatively more associated with breast malignancies, although its expression is also reported in other cancers as well. Likewise, the over activity of lysophosphatidic acid is highly associated with ovarian cancers. It has been reported earlier that the expression of Ang 1 is negatively associated with the adipose tissue weight. Contrarily, Ang 2 expression is increased in the adipose tissue where it modulates adipose tissue vasculature [31]. Likewise, ANGLP 2 (along with other ANGLPs) is a pro-inflammatory cytokine that is released from the adipose tissue to activate macrophages. The tumor angiogenesis in the tumor supportive microenvironment is attributed to an over-expression of Ang 1, 2, 3 and 4 as well as many ANGLPs in different cancer types.
Owing to the complexity of cancer pathogenesis, several different theories have been postulated in past few years to relate these mediators with cancer initiation and progression. However, the certainty regarding the extent of involvement of these suggested disease mechanisms may incite another discussion that does not come under the scope of this article. For instance, a relatively higher prevalence of malignancies such as the breast, colon, pancreas, and prostate in obese population is already established. Nonetheless, all obese people do not suffer from these cancers in their entire lifespan despite of the fact that their cells are exposed to a microenvironment rich in obesity mediators. We hereby describe the suggested involvement of these mediators with malignancies in a concise manner.
Leptin
Leptin is a peptide adipokine that plays a significant physiological role in controlling the total mass of adipose tissue to maintain homeostasis. Its physiological significance can be judged by the fact that the genetic silencing of leptin or its receptor have resulted into serious pathological defects such as metabolic derangements, infertility, immune deficiencies and diabetes in knockout mice [32]. Moreover, it affects several cell populations under physiological conditions, including the immune cells, neurons, pancreatic β cells, adipocytes and endothelial cells that suggests a broader physiological role of leptin compared with other adipokines. Once released from the adipocytes, leptin elicits its hunger-suppressing effects by inhibiting neuropeptide Y and anandamine via activating its receptors in the lateral hypothalamus. Under the normal physiological conditions, it regulates proliferation [33], invasion [34] and the absorptive properties [35] of colon cells. Owing to its higher serum concentration in obesity, leptin is one of the most studied adipokines in the progression and metastasis of breast cancer. Higher leptin levels are frequently reported in breast, colon and pancreatic cancers [36,37]. Leptin levels are also correlated with different breast cancer types. For instance, its levels were found higher in patients with ductal carcinoma in situ as compared to the invasive breast cancer patients where adiponectin was significantly higher than leptin [38]. Although a previous study claimed either no or even a negative correlation between the serum leptin levels and pre-menopausal breast cancer incidence [39,40], the interrelationship between leptin serum levels and breast cancer risk in post-menopausal women is well-established. In addition to the adipose, leptin is also synthesized and released in significantly higher concentrations from glioblastoma cells that majorly accounts for the morbidities associated with glioblastoma [41]. It elicits its biological function through the leptin receptor B (ObRB) via activating PI3K, JAK2/STAT3 and ERK1/2 signaling. The recent evidence not only supports the possible role of leptin in the initiation of cancer, but it has also been confirmed to promote the invasion and metastasis of cancer cells by activating EMT, inhibiting immune checkpoints and bolstering angiogenesis.
The impact of leptin treatment on cancer cell proliferation varies greatly from one type of cells to the other. It exhibited anti-proliferative effect on Chang liver cells, where the proliferation rate in the leptin-treated cells was found significantly lower than the control [42]. Contrarily, it induced proliferation in several cell lines such as the hepatocellular carcinoma cells HepG2 [43], Huh7 [44] and the ovarian adenocarcinoma cells OVCAR3 [45]. When the HepG2 and OVCAR3 cells were exposed to different leptin concentrations (0–200 ng/ml), a significantly higher population of cells crossed the G0–G1 phase in a dose dependent manner and were found in S and G2-M phase [43,45]. These findings suggested a direct impact of leptin on DNA synthesis and mitotic division. Leptin triggers JAK/STAT, p44/p42, Erk1/2 and Akt signaling where STAT3 plays pivotal role in exhibiting its proliferative and carcinogenic effects [44,46–49]. The STAT3 role in leptin signaling is further attested by the fact that the inhibition of STAT3 had shown to promote cell cycle arrest, apoptosis and impede tumor invasion [47,50,51]. Moreover, a differential pattern of gene expression was observed in the leptin exposed tissue samples from patients suffering from colorectal cancers where a higher expression of STAT3, Akt1 and MCL-1 was observed in the earlier non-metastatic stages whereas a highly expressed CCND1 and VEGFC genes were observed in the advanced metastatic stages [52]. The role of STAT3 in leptin induced cell proliferation and metastasis is described in Figure 3. It has been suggested that STAT3 mediated mitogenic effects are shared by the signaling through leptin and IL6Rα receptors. A decline in IL6Rα receptors on hepatocytes has boosted the expression of leptin receptors in IL6Rα deficient mice, suggesting the direct involvement of leptin-IL6 interplay in the tumorigenesis of hepatocellular carcinoma [53]. Further, leptin signaling was reported to maintain the stemness in triple negative breast cancer cells via upregulating the self-renewal transcription factors such as SOX2, OCT4 and NANOG via STAT3 signaling [54,55].
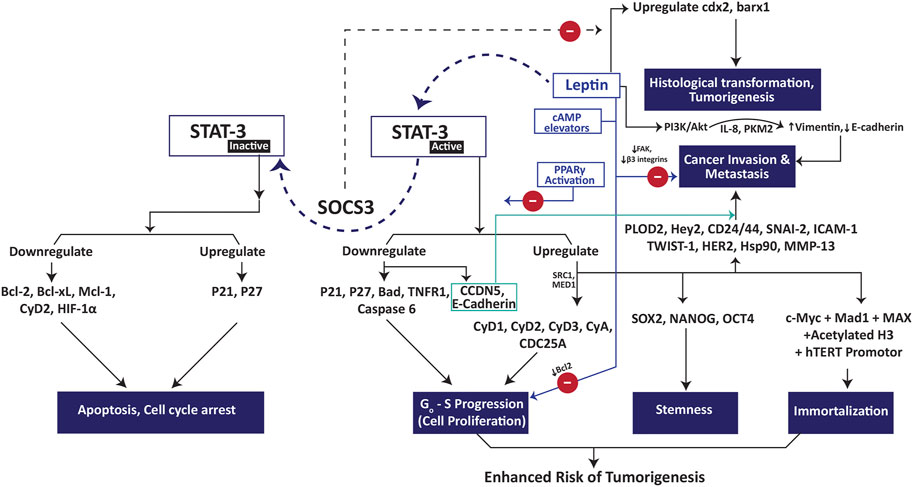
FIGURE 3. Role of STAT3 in leptin-induced tumorigenesis, cancer cell proliferation and metastasis. Abbreviations: Bad Bcl-2 associated agonist of cell death; Bcl-2 B-cell lymphoma-2; Bcl-xL B-cell lymphoma 2 extra large; CyA cyclin A, CyD1 cyclin D1; CyD2 cyclin D2; CyD3 cyclin D3; Hey2 hairy/enhancer-of-split related with YRPW motif protein 2; HIF-1α hypoxia inducible factor-1 alpha; hTERT human telomerase reverse transcriptase; MAD-1 mitotic arrest deficient-1; MAX myc-associated factor X; Mcl-1 myeloid cell leukemia 1; MMP13 matrix metallopeptidase 13; PLOD2 procollagen-lysine 2-oxoglutarate 5-dioxygenase 3; SOCS3 suppressor of cytokine signaling 3; STAT-3 signal transducer and activator of transcription 3; TNFR1 tumor necrosis factor receptor 1.
The proliferative properties of leptin were reported to get influenced by several cofactors. For instance, leptin has increased proliferation of colon cancer cells in vitro however, it failed to support the tumor growth in vivo [56]. Likewise, it induced proliferation in androgen-sensitive prostate cancer cells (LNCaP) but failed to influence the proliferative rate of androgen-insensitive PC3 and Du-145 cells [57]. Besides, it inhibited the cell migration and invasion in the mentioned androgen-insensitive cells by activating MAPK pathway, a change that was not observed in androgen-sensitive cells. Further, it activated STAT3 signaling in both types of the cells but with a significant difference. In androgen-insensitive cells, the STAT3 signaling was observed in the earlier phase of the cell cycle with a gradual decline in the later phases. Hence, the leptin-induced STAT3 remained unable to trigger proliferation and precipitated in the blockage of invasion and cell migration in PC3 and Du-145 cells. However, in the androgen-sensitive LNCaP cells, a relatively delayed but persistent activation of STAT3 was observed that boosted the proliferative rate in these cells [57]. Interestingly, leptin associated cell proliferation and migration via STAT3 signaling are reversed if cAMP levels are elevated. For instance, the co-treatment of MDA-MB231 cells with leptin and cAMP elevating agents (8-pCPT-2′-O-Me-cAMP, 8-Br-cAMP) blocked the proliferative effect of leptin and induced apoptosis in cancer cells by downregulating Bcl2 [58]. Such elevation of cAMP also blocked leptin-induced cell migration in MDA-MB231 cells by downregulating FAK and β3 integrins via cAMP/PKA and cAMP/Epac signaling [59]. Moreover, the activation of PPARγ by rosiglitazone has also abrogated leptin induced cell proliferation and metastasis by blocking the activation of MAPK/STAT3/Akt signaling in both the in vitro and in vivo experiments [60].
In ER negative breast cancer cells (MDA-MB-231 and BT-549), leptin induced the expression of oncogenic enzyme sphingosine kinase 1 (SK-1) via Src family kinase (SFK) and Erk1/2 signaling. However, such upregulation of SK-1 was not observed in ER positive breast cancer cells [61]. Importantly, the role of the suppressor of cytokine signaling (SOCS) should also be considered while discussing the proliferative properties of leptin. The upregulation of SOCS3 inhibits ERK and Jak2 activation [62] and attenuates Akt signaling [63]. The persistent stimulation of leptin receptors precipitates in the higher expression of SOCS3 as a negative feedback mechanism [64]. Moreover, p38-MAPK signaling also results into an upregulation of SOCS3 [65]. Hence the anti-proliferative effect of leptin in H4IIE cells could possibly be attributed to the SOCS3 mediated attenuation of the mitogenic pathways [66]. Contrarily, the irrepressible leptin signaling in SOCS3 deficient mice has exacerbated tumorigenesis in gastric and intestinal epithelium by upregulating cdx2 and downregulating the transcription activities of barx1 [67]. Aside from the mentioned factors, the expression of both methionine adenosyltransferase 1α (MAT 1α) and MAT 1β is necessary for the mitogenic effect of leptin in hepatocellular cancer cells [68].
Long-term exposure of cells to leptin results into the upregulation of human telomerase reverse transcriptase (hTERT) expression by STAT3 mediated binding of c-Myc, Mad1, Max and acetylated H3 with hTERT promotor [69]. The above notion is further confirmed by the fact that leptin-induced cancer cell proliferation is inhibited by 1 alpha, 25-dihydroxyvitamin D3 through miR-498 mediated downregulation of hTERT [70]. hTERT elongates the telomere length to prevent the cells entering senescence after several cell divisions, a process that is generally termed as immortalization. Together with the proliferative properties of leptin, this immortalization of cells after being exposed to leptin for longer may lead to the basis of tumorigenic transformation of cells. STAT3 activates glycoprotein 130 (gp130) subunit of cytokine receptors that further promotes the progression of cells from Go to S phase through upregulating Cyc D1-3, A, cdc25A and downregulating P21, P27 [47,71]. Besides, leptin is reported to induce the expression of Notch1-4, JAG1 and DLL4 in pancreatic cancer cells [72]. The resultant activation of Notch signaling further accounts for the leptin-induced cell proliferation in cancer. More recently, the supportive role of ribosomal protein S27/Metallopanstimulin-1 (MPS-1) in cancer proliferation and invasion is reported in different cancer cells, including glioma [73], colorectal [74] and gastric cancer cells [75]. Leptin is found to upregulate MPS-1 in colorectal cancer cells that further promoted the cell survival and proliferation via activating JNK/c-JUN signaling [74]. Moreover, the role of insulin growth factor-1 (IGF-1) in promoting cancer cell proliferation and invasion is well documented [76]. Leptin treatment had significantly upregulated IGF-1 in breast cancer cells via activated protein-1 (AP-1) transcription factor [77]. The silencing of the three MAPK mitogenic pathways i.e., JNK1/2, p38 MAPK and Erk1/2 abrogated IGF-1 upregulation suggesting the direct involvement of these signaling pathways in leptin-induced IGF-1 upregulation. It is worth mentioning that the extracellular cell survival factors, such as the nutrients, trigger cell proliferation by upregulating JAK/STAT, Erk and PI3K/Akt signaling. Leptin does not appear to halt the proliferation of cells directly. Instead, it exhibits its anti-proliferative effect by attenuating these mitogenic signaling pathways. The evidence to support this notion comes from the findings of Thompson et al. 2011 wherein leptin failed to affect the proliferation of rat H4IIE cells in the serum depleted medium [66]. However, it showed a significant inhibitory effect on these cells in the presence of serum rich medium by upregulating p38-MAPK signaling.
Leptin supports cancer cell survival by inhibiting the apoptotic pathways through downregulating Bad, tumor necrosis factor alpha receptors 1 (TNFR1) and caspase 6 expression [45]. It also supports growing tumor by promoting lymphangiogenesis via upregulating proangiogenic proteins VEGF, IL-1β and leukemia inhibitory factor (HIF) in cancer cells [78–80]. The upregulation of VEGF was found to involve an elevated expression of HIF-1α, NF-κB [81], Notch and IL1 [82] signaling. Further, the mentioned upregulation of HIF-1α involved both the canonical and non-canonical pathways, whereas the NF-κB regulation was mainly attributed to the non-canonical pathway [81]. It is a worth-mentioning finding here that leptin at higher dose induces senescence and Go-S phase arrest in chondrogenic progenitor cells (CPC) by inhibiting Sirtuin 1 pathway and upregulating p57/p21 expression [83]. These changes impart osteogenic properties to CPC that may account for the pathophysiology of obesity derived osteoarthritis. The adipocytes released leptin and IL-6 to promote metastasis in breast cancer cells by upregulating PLOD 2 expression via JAK/STAT and PI3K/AkT signaling [84]. In colon cancer cells (HCT116), it promoted Rac-1 and Cdc25 induced invasion, lamellopodia formation and metastasis by activating PI3K and Src kinase pathway [85]. The secretion of leptin is significantly increased from the obesity modified adipose stem cells (obASC) [86] that further promotes EMT in cancer cells, paving a way towards cancer metastasis. Leptin promoted the phosphorylation and inactivation of GSK3β via STAT3 mediated Akt phosphorylation [87]. Moreover, leptin also upregulated metastasis associated protein 1 (MTA1) in breast cancer cells that further contributed to the inactivation of GSK3β via Wnt1 signaling. The mentioned GSK3β inactivation abrogates the formation of its complex with liver kinase B1 (LKB1) and axin, precipitating in the cytosolic accumulation and nuclear translocation of β-catenin. Once inside the nucleus, β-catenin induces the expression of a set of genes including Cyc D1, Cyc D2, Myc, WISP1 (CCN4) and WISP2 (CCN5) to execute EMT. The blockage of Wnt/β-catenin signaling completely abrogated the leptin induced proliferation in breast cancer cells, suggesting its direct involvement in cancer cell proliferation [88]. Interestingly, leptin downregulated the expression of CCN5 in breast cancer cells MCF-7 and ZR-75-1 cells via activating JAK/STAT and Akt pathways [89]. Such downregulation of CCN5 did not affect cell proliferation, but enhanced cell viability, invasion and EMT.
Leptin upregulates the expression of Notch1 by transactivation of epidermal growth factor receptor [90]. This is followed by the recruitment of Notch-1 intracellular domain on the promoter region of survivin. These findings are further supported by the results of another study wherein leptin promoted breast cancer metastasis in mice model by upregulating the levels of Notch 3, Jag1 and survivin [91]. It also enhanced the expression of Notch1, 1-4, JAG1, DLL4 and surviving in pancreatic cancer cells [72]. The resultant upregulation of survivin promotes the migration and invasion of cancer cells, suggesting a direct role of leptin-Notch-EGFR axis in leptin induced metastasis. Interestingly, an increased release of leptin from the pancreatic cancer cells was found to modify the tumor microenvironment further by upregulating the expression of leptin receptors in an autocrine/paracrine fashion [72]. Such upregulation of leptin receptors in also reported in breast cancer cells where the leptin-treatment increased the expression of AdipoR2 receptors in MCF-7 cells [92].
Hey2 (Hairy/enhancer of split related with YRPW motif protein 2) is a transcription factor that is recently reported to play a critical role in cell proliferation and metastasis in hepatocellular carcinoma [93]. Although its expression in hepatocellular carcinoma cells is not linked with leptin elicited effects, its expression along with other pancreatic cancer stem cell markers (including Oct-4, CD133, CD24/44 and ALDH) is significantly increased in leptin treated pancreatic cancer cells [72]. The overexpression of CD24 reduces E-cadherin but at the same time upregulates N-cadherin to accelerate cancer invasion [94]. Likewise, the overexpression of CD44 confers aggressive and distant metastasis in cancer cells [95]. In lung and breast cancer cells, leptin treatment has amplified the levels of soluble ICAM-1 by upregulating ERK, GSK3αβ, JAK1/2, STAT3 and focal adhesion kinase (FAK) signaling [96] without affecting the expression of cell surface ICAM-1. Such elevation of soluble ICAM-1 in cancer cells promotes bone metastasis by inducing osteoclast-like cell formation [96]. It promoted cell migration and metastasis in gastric cancer cells (AGS, MKN-45) by enhancing the expression of ICAM-1 (both soluble and surface) via Rho/ROCK signaling [97]. The stromal cell derived leptin is reported to support cancer metastasis in estrogen receptor positive [98] and triple negative [99] breast cancer cells by enhancing EMT via upregulating SERPINE 1, IL-6, MMP-2, SNAI 2, TWIST 1, vimentin and downregulating E-cadherin [100]. The leptin induced EMT in breast cancer cells was reported to involve an upregulation of IL-8 [101] and pyruvate kinase M2 (PKM2) [100] via PI3K/Akt signaling. These findings were further confirmed by the fact that the silencing of leptin signaling in highly metastatic MDA-MB-231 cells had resulted into MET via upregulating E-cadherin and downregulating vimentin [54]. Leptin also induced EMT in lung cancer cells by downregulating E-cadherin, keratin and upregulating vimentin and the transcription factor ZEB-1 via Erk signaling [102].
Leptin exhibits a dose and time dependent activation of mTORC and its downstream protein p70S6 kinase that precipitates into an increase in cell proliferation and upregulation of cyclooxygenase 2 (COX2), transforming growth factor-beta (TGF-β), monocyte chemoattractant protein 1 (MCP-1) and cytokine induced neutrophil chemoattractant-1 (CINC-1) [103]. Likewise, leptin upregulates the expression of TNF-α in macrophages [104]. The activated p70S6 kinase is reported to boost cancer metastasis and invasion through IL-6 mediated EMT in squamous cell carcinoma [105]. Moreover, the upregulation of TGF-β, TNF-α and COX-2 promote EMT in different cancers that results into the poor prognosis of cancer therapy [106–110]. The upregulation of heat shock protein 27 (hsp27) in leptin treated MCF-7 cells may also account for the leptin-induced metastasis [111], since the role of hsp27 is well documented in suppressing apoptosis and promoting cancer cell invasion and metastasis [112]. Leptin also promoted cancer growth and progression by STAT3-mediated upregulation of MMP-13 [113], HER2 and hsp90 [114] in pancreatic and breast cancer cells respectively.
It is recently reported that the treatment of triple negative breast cancer cells with leptin has upregulated the expression of oncogenic and chemotherapy-resistant genes including ITGB3 (integrin β3), LL22NC03, ABCB1 (p-glycoprotein 1), WNT4 (WNT4 protein), TBC1D3 (TBC1 domain family member 3E/3F), ADHFE1 (alcohol dehydrogenase, iron containing 1) and RDH5 (11-cis retinol dehydrogenase) [115]. Likewise, it promoted tumorigenesis in ovarian epithelial cells by enhancing the expression of histone deacetylase (HDAC) class I and II [116]. It is thereby suggested that leptin is not only involved in tumorigenesis, it also contributes to the resistance of cancer cells against chemotherapy [115,117]. For instance, the cytotoxic potential of 5-fluorocracil is countered by leptin by attenuating the pro-apoptotic proteins Bax and caspase 3 [118]. It is also important to state that the major tumorigenic aspects of cancer biology such as the immunosuppression, malignant transformation, angiogenesis, and metastasis involve a complex exosomal cell-to-cell communication between cancer cells [119]. Leptin is reported to enhance such exosomal cell communication in estrogen positive MCF-7 and triple negative MDA-MB-231 breast cancer cells via hsp90 mediated post-transcriptional increase in the levels of tumor susceptibility gene 101 (Tsg 101) protein [120].
In addition to its role in cell proliferation and metastasis, leptin also supports the survival of cancer cells under nutrient-deficient tumor microenvironment by promoting autophagy. It is reported to induce autophagy in breast cancer and hepatocellular carcinoma cells by enhancing the expression of LC3II, Atg5, Beclin1 and downregulating p62 expression [121,122]. Such induction of autophagy by leptin involved the activation of p53 and AMPK/FoxO3A pathway. Leptin induced an upregulation of p53 in HepG2 and MCF-7 cells without affecting the p53 mRNA levels [123]. Instead, the upregulation was found to involve the regulation of p53 ubiquitination via ubiquitin specific protease-2. In breast cancer cells, the expression of autophagy genes was found to involve estrogen receptors ERα signaling, since the inhibition of ERα abrogated leptin-induced autophagy [121]. The leptin signaling also suppresses mitochondrial respiration in cancer cells, an act that further promotes invasion, metastasis and enhances the survival of these defiant cells under hypoxic conditions [124,125].
Resistin/Adipose Tissue Specific Secretory Factor (ADSF)/FIZZ3
The expression of resistin was found significantly higher in estrogen negative breast cancer patients and was reported as a potential biomarker for early cancer detection [29]. The positive correlation between serum resistin levels, cachexia and metastasis in gastroesophageal cancers affirms its direct involvement in cancer pathogenesis [27]. Further, its higher expression is found associated with poor prognosis, nodal metastasis, and advanced cancer stage in colorectal cancer patients [126]. It is important to state that its expression was negatively associated with high density lipoprotein cholesterol (HDL-C) in colorectal cancer patients because of a possible interaction of its release with HDL-C [127].
Mechanistically, resistin was found to promote invasion, migration and cellular protrusions in cancer cells by inducing the reorganization of F-actin filaments [128]. In the breast and ovarian cancer cells, resistin upregulated the expression of SNAIL, SLUG, ZEB1, TWIST1, fibronectin, vimentin and downregulated claudin-1 and E-cadherin via AXL tyrosine kinase receptor signaling [128–130]. Further, it induced the adhesion molecules ICAM-1 and VCAM-1 in hepatocellular carcinoma cells SK-Hep1 [131] and colorectal cancer cells HCT-116 [132] through NF-κB signaling. These adhesion molecules tend to attach the cancer cells with the HUVECs, a process that may account for intravasation. It also potentiated the nuclear translocation of SNAIL to induce EMT, an important pre-requisite for SNAIL induced mesenchymal transition. Further, the resistin induced EMT was completely abrogated after silencing adenyl cyclase associated protein 1 (CAP-1), suggesting a direct involvement of CAP-1 in resistin induced metastasis [128]. Moreover, the significant role of CXC chemokine receptor 4 (CXCR4) in lymphangiogenesis and cancer metastasis is well documented. Resistin is reported to enhance the expression of CXCR4 in gastric cancer cells via TLR4 receptor signaling [133]. Further, the resistin induced upregulation of CXCR4 is abrogated by blocking p38-MAPK and NF-κB pathway, suggesting the involvement of these signaling proteins in resistin induced CXCR4 upregulation.
The resistin treatment enhanced stemness in cancer cells by upregulating the stemness markers aldehyde dehydrogenase-1 (ALDH1), CD44, Oct4, Nanong and Sox2 and downregulating CD44 cell surface expression [130,134]. It also promoted cell proliferation in gastric cancer cells by upregulating the expression of hTERT gene [135]. Apart from its impact on cell proliferation and EMT, it also exhibits chemotherapy resistance. For instance, it was found to exert doxorubicin resistance in breast cancer cells by blocking the chemotherapy-induced apoptosis via activating STAT3 signaling [134]. Likewise, the efficacy of dacarbazine was also attenuated by resistin through upregulating caveolin-1 [136] in melanoma cells [136]. It also induced accumulation of LC3 in breast cancer cells to support their survival under drastic chemotherapeutic conditions via activating autophagic pathways [137]. It was found that the treatment with resistin culminates into autophagy by upregulating BECN1, LAMP1, LC3B-II and downregulating SQSTM1. Such activation of autophagic pathways was completely abrogated by blocking the JNK and AMPK/mTOR/ULK1 signaling [137], suggesting the direct involvement of these two pathways in resistin induced autophagy.
Visfatin
Visfatin is reported to induce cell proliferation in different cancer cells such as melanoma and breast cancer cell [138–140]. Like leptin, visfatin also stimulates a number of cell signaling pathways that support cell proliferation and survival, such as the PI3K/mTOR, ERK1/2, p38 MAPK, JAK/STAT and JNK [141–145]. This proliferative effect of visfatin is conferred by its ability to stimulate notch 1 signaling through activating the p65 unit of NF-κB [146]. It induces cell survival in melanoma cells by inhibiting p53 dependent apoptosis [140]. The intensity of cancer invasion and metastasis in gastric [147] and colorectal cancer patients [148] was found correlated with their higher serum visfatin levels. Its high levels were also found correlated with the ability of small cell lung cancer (SCLC) cells to metastasize into brain via transendothelial migration across the blood brain barrier [149]. Such migration of SCLC cells across the brain endothelial cells was found to involve upregulation of chemokine ligand 2 (CCL2). It is worth mentioning here that the CCL2-CCR2 signaling plays a crucial role in both intravasation and extravasation through the recruitment of CCR2 positive myeloid cells [150]. These myeloid cells help the recalcitrant CCL2 positive cancer cells to enter into blood/lymph vessels from their site of origin and then extravasate to a new location. Likewise, it also imparts motility in cancer cells via NF-κB and IL-6 signaling [151] and promotes EMT by facilitating the nuclear transport of the transcription factors SNAIL and β-catenin [148]. Moreover, it also inhibits its binding with GSK3β in cytosol to prevent its’ ubiquitination [148]. The ascites-driven visfatin was found to promote metastasis in ovarian cancer cells by inducing lamellipodia and filopodia via Rho/ROCK signaling [149]. In non-small cell lung cancer cells, it induced resistance to the chemotherapeutic effect of doxorubicin by Akt induced upregulation of multidrug resistance protein-1 (MRP-1) [152].
Omentin
Since omentin-1 (intelectin-1) is the major adipokine from the visceral fat (that is accumulated in the omentum), its emerging significance in the metastasis of ovarian and gastric cancers is well reported [153,154]. The omentum is covered with a thin layer of mesothelial cells. To metastasize, the cancer cells have to bind and penetrate through the mesothelium. At first, the anti-metastatic potential of omentin-1 tries to block the penetration of the cancer cells through mesothelium, probably by involving lactotransferrin (LTF). The interaction of LTF with its receptor is crucial for the up-regulation of matrix metalloproteinase 1 (MMP1). By inhibiting LTF binding with its receptor LRP-1 (low intensity lipoprotein receptor related protein 1), omentin-1 attenuates the upregulation of MMP1 in ovarian cancer cells [153]. As MMP1 plays a significant role in invasion of cancer cells through extracellular matrix, its down regulation in ovarian cancer cells accounts for the anti-metastatic potential of omentin-1 [153]. Likewise, omentin-1 blocks the PI3/AKT mediated up-regulation of NF-κB signaling [154]. Since NF-κB suppresses the expression of hepatic nuclear factor 4 alpha (HNF4α), omentin-1 upregulates its expression in gastric cancer cells via attenuating NF-κB signaling [154]. HNF4α blocks the metastatic effects of β-catenin by inhibiting its nuclear translocation, a mechanism that accounts for the anti-metastatic effect of omentin-1 in gastric cancer cells [154]. However, the cancer cells escape omentin-1 mediated anti-invasive barrier by downregulating its expression in mesothelial cells through TNF-α signaling [155]. Likewise, the ovarian cancer cells upregulate fibronectin in the mesothelial cells of omentum via TGF-β dependent Rac1/SMAD signaling. Such upregulation promotes invasion and metastasis in these cells, departing the anti-metastatic effect of omentin-1 [156].
Adiponectin
As mentioned earlier, adiponectin exhibits promising anti-proliferative and pro-apoptotic properties. Such potent anti-cancer potential of adiponectin is also reflected from the fact that the expression of adiponectin receptor 1 and 2 is downregulated in highly metastatic cancers. For instance, the tissue samples from colorectal cancer patients with nodal metastasis exhibited a significant downregulation of adiponectin receptors 1 and 2 as compared to the normal tissue samples [157]. Such downregulation of adiponectin receptors could be considered as a survival mechanism in these metastatic cancer cells to evade the anti-proliferative and pro-apoptotic effect of adiponectin. Adiponectin attenuates cancer metastasis by upregulating the expression of tumor suppressor protein LKB1 [158]. LKB1 inhibits mTOR complex 1 (mTORC1) by phosphorylating tuberous sclerosis proteins 2 (TSC2) via phosphorylating AMP-activated protein kinase (AMPK) [158,159]. It is worth mentioning here that the blockage of mTORC1 by LKB1 does not only depend upon TSC2 activation as it is equally suppressing mTORC1 in TSC2 lacking cells. Such TSC-2 independent blockage of mTORC1 by activated AMPK involves a direct phosphorylation of raptor protein on two distinct positions i.e., serine 722 and serine 792 which precipitates into the binding of raptor with 14.3.3 protein [160]. By blocking mTORC1 via LKB1, adiponectin suppresses the activation of its downstream protein P70SK [158] which is a key regulator of cell invasion and metastasis in many different types of malignancies [161,162]. Importantly, such antimetastatic effect of adiponectin is not observed in ER positive breast cancer patients. In such patients, the EAα-LKB1 interplay attenuates the LKB1/AMPK mediated inhibition of mTORC1 with a subsequent promotion of invasion and metastasis in ER positive breast cancer cells [163]. Hence, the LKB1 mediated blockage of p70SK by adiponectin may account for its anti-metastatic properties. Moreover, adiponectin inhibits the intracellular accumulation of β-catenin via blocking GSK3β phosphorylation [164]. Such inhibition of β-catenin may account for the anti-metastatic potential of adiponectin.
Adiponectin inhibits the proliferation of cancer cells by downregulating STAT3 and upregulating SOCS3 [165,166]. Further, its inhibitory effect on the proliferation, invasion and metastasis of cancer cells could also be attributed to the attenuation of PI3K/Akt [167], Erk 1/2 [168–170], mTOR/S6 [171] and NF-κB signaling [172]. It also downregulates the expression of Cyc D1 via blocking the Akt-mediated phosphorylation of GSK3β [173]. Likewise, it downregulates c-Myc, Cyc D, Bcl2 in cancer cells with an upregulation of p53, p21, p27 and Bax [170,174,175]. In colon cancer cells, adiponectin treatment halted the proliferation by activating AMPK signaling. Further, it also suppressed the expression of SREBP1c that contributed to its antiproliferative effect in colon cancer cells [175].
Angiopoietin
The impact of Ang 1 expression on cancer metastasis stands dubious since a number of studies have reported “conflicting’’ results regarding its pro-metastatic or anti-metastatic potential. Owing to the fact that the cancer cells tend to invade the surrounding tissue by breaking their cell-cell and cell-ECM attachments, the mediators that strengthen such intercellular junctions suppress such invasion and metastasis. Kitajima and colleagues have recently reported that the overexpression of Tie2 receptors in oral squamous cell carcinoma bolster the cell-cell and cell-matrix attachment [176]. In a subsequent study, Kitajima and colleagues claimed a significant enhancement in the tie2-induced cell-cell and cell-matrix attachment by Tie2/Ang 1 interaction [177]. They also suggested a negative correlation between nodal metastasis and Tie2 and/or Ang 1 expression [177]. In another parallel study, the Ang 1 knockout mice showed significant increase in the metastasis of breast and melanoma cells to the lung tissue without affecting the growth of the primary tumor [178]. Since the colonization of cancer cells in lungs was significantly increased in Ang 1 knockout mice after direct intravenous injection of melanoma cells, it was suggested that Ang 1 deficiency might have promoted the last stage of metastasis i.e., the extravasation in the knockout mice [178]. Likewise, Ang 1 and vasculoid (Tie2 agonist) blocked the trans-endothelial migration of colon, breast and renal cancer cells in vitro [179]. Moreover, vasculoid also delayed the spread of breast cancer cells to lungs [179]. In contrast, the expression of Ang 1 in cancer cells through adenoviral vector-mediated transfection has been reported to induce cancer metastasis via overexpression of β1 integrin and CD44 [180,181]. The expression of β1 integrin and CD44 was found to increase the attachment of gastric cancer cells with the extracellular matrix that might have increased their invasive potential [180]. The findings of Holopainen and coworkers suggested that Ang1 expression enhances vascular enlargement, without significantly affecting vascular density. Such enlargement of blood vessels assists in the extravasation of cancer cells by promoting their dissemination in blood [181]. Summarizing, Ang1 exhibits a dual role in cancer metastasis. In the early stages, it exerts an inhibitory effect on cancer invasion by strengthening the cell-cell junctions via Tie2 receptors. However, once the cancer cells succeed in invading the extracellular matrix via EMT, the Tie2/Ang 1 signaling promotes the trans-endothelial migration of these cells via bolstering their attachment with endothelial cells, a prerequisite for both intravasation and extravasation. It is important to mention here that the pulmonary metastasis in Lewis lung carcinoma cells (LLCC) as well as in the TA3 mammary carcinoma cells was found to involve VEGF and Ang 1 induced angiogenesis [182]. VEGF and Ang 1 promote angiogenesis in these carcinoma cells through ERK 1/2 and Akt pathways. Ang 3 blocks pulmonary metastasis in LLCC and TA3 carcinoma cells by suppressing the expression of both VEGF and Ang 1 [182]. The binding of Ang3 with the cell surface through heparan sulfate proteoglycans was found mandatory to inhibit Ang 1 and VEGF mediated angiogenesis and metastasis in LLCC and TA3 cells [182].
Ang2 is reported to promote cancer metastasis by activating EMT in breast cancer [183] and oral squamous cell carcinoma [184]. In Tie-2 deficient cells, Ang 2 promotes the activation of ILK, GSK3β and Akt via its binding with α5β1 integrin [183]. This activation of ILK and GSK3β induces SNAIL expression, downregulates E-cadherin and promotes EMT to induce metastasis [184]. An estrogen depletion therapy in ER + breast cancer patients may instigate cancer recurrence and metastasis via Ang 2 involvement. Ang 2 exhibits such revival of dormant ER + breast cancer cells in the vascular niche of bone marrow either by directly inhibiting Ang 1-Tie2 signaling, or via stimulating surface integrin β1 receptors in these dormant cancer cells [185].
Angiopoietin Like Proteins (ANGLP)
Several reports have correlated the expression of ANGLP 2 with cancer metastasis. For instance, the overexpression of ANGLP 2 was found associated with the lymph nodal metastasis in the clinical samples from the surgically treated non-small cell lung cancer patients [186]. Likewise, ANGLP 2 was reported to promote carcinogenesis and metastasis to distant organs in mice by promoting EMT [187]. The expression of transcription factors such as the nuclear factor of activated T cells (NFATc), activating transcription factors (ATF) and c-JUN in aggressive tumor cells result into the upregulation of ANGLP 2 [188]. Further, HIF-1α is recently reported to upregulate ANGLP 2 expression in hypoxic osteosarcoma cells [189]. The upregulation of ANGLP 2 further promotes motility and invasion in the cancer cells in either an autocrine or a paracrine fashion [188]. ANGLP 2 promotes metastasis though facilitating angiogenesis as well as glycolysis via upregulating VEGFA, Ang 2 and hexokinase 2 [189]. Further, it promoted intravasation in osteosarcoma cells via upregulating p38 MAPK signaling, MMP as well as integrin α5β1 [190].
Earlier, ANGLP 4 was reported to inhibit metastasis in melanoma cells by inhibiting cancer cell motility and reducing vascular permeability to the invading cells [191]. However, the subsequent studies have reported the opposite findings. ANGLP 4 is found to promote lung metastasis in breast cancer cells via blocking endothelial to endothelial cell interactions [192]. The inhibition of these endothelial cell interactions is necessary for the trans-endothelial migration of cancer cells during the intravasation and extravasation. Since TGFβ was found to upregulate the expression of ANGLP 4 in breast cancer cells, the lung metastasis induced by TGFβ was found to involve ANGLP 4 mediated increase in vascular permeability [193]. Likewise, HIFα also exhibited its metastatic potential by upregulation of ANGLP 4 [192]. ANGLP 6 is found within the blood vessels of the colorectal cancer patients. It is reported to bind with tumor cell specific integrin α6 and E-cadherin on the surface of the tumor cells that precipitates into the liver metastasis of colorectal carcinoma cells [194].
Chemerin
Chemerin exerts its biological functions through G protein coupled receptor 1 (GPR 1) and chemokine like receptor 1 (CMKLR 1) [30]. The expression of chemerin and its receptors are highly upregulated in adipose tissue and the recent research has reported a positive correlation between circulating chemerin levels and obesity [195]. Its role in promoting the invasion and metastasis of cancer appears to be dubious, since it exhibits both tumor suppressive and promoting effects that depends upon the type of the disease. For instance, it is reported to exhibit cancer suppressive effect in hepatocellular carcinoma [196]. The downregulation of chemerin expression in malignant adrenocortical carcinoma [197] and acute myeloid leukemia [198] could be assumed as a survival tactic of the cancer cells to evade the immunosuppressive effect of chemerin. Contrarily, its expression is positively associated with malignancy in non-small cell lung carcinoma [199] and gastric cancers [200]. The complexity of chemerin role in breast cancer could be explained by the conflicting findings of two recent studies. Pachynski and coworkers have reported that the expression of chemerin in the breast tumor microenvironment suppresses the progression of disease by promoting the infiltration of immune cells [117]. Contrarily, El-Sagheer et al has reported a comparatively higher expression of chemerin in malignant tissue, suggesting a significant association of chemerin with poor prognosis in breast cancer patients [201].
First, we focus on the cancer suppressive potential of chemerin that could mainly be attributed to its immunomodulatory and chemoattractant potential. Binding of chemerin with its receptors CMKLR1 and GPR1 has reported to induce receptor internalization and deactivation [202]. Such internalization of the G-protein coupled receptors further recruits β-arrestin 1 and 2 with a subsequent activation of β-arrestin 2 dependent ERK1/2 activation [202]. It is worth mentioning here that the role of β-arrestin 2 in cancer stands dubious till date. Although a few reports have suggested its tumor promoting potential in renal [203] and breast cancer [204], a considerable data suggests that the activation of β-arrestin 2 inhibits tumorigenesis. By acting as a corepressor of androgen receptors, β-arrestin 2 downregulated FOXO1 with subsequent inhibition of cell proliferation in prostate cancer cells [203,205]. Moreover, its depletion/less expression has resulted into aggressive cancer invasion with poor prognosis in non-small cell lung cancer [206] and hepatocellular carcinoma [207]. Importantly, the β-arrestin mediated activation of ERK1/2 promotes the cytosolic functions of ERK1/2 by impeding its nuclear translocation [208]. Contrarily, the AMP-PKA mediated activation of ERK1/2 promotes the nuclear translocation of ERK1/2 with subsequent expression of proliferative genes. Hence, the chemerin-β arrestin signaling may contribute to its tumor suppressive potential. Besides, chemerin dependent activation of β-arrestin 1 and 2 may account for the dubious role of chemerin in different types of cancers as mentioned above.
Chemerin is reported to upregulate the expression of serum response factor (SRF) in lymphocytes and gastric carcinoma cells through Rho/ROCK signaling [209]. Importantly, the overexpression of SRF was found associated with poor prognosis and shorter survival rates in patients with gastric carcinoma [210], bone and prostate malignancies [211]. SRF exerts its cellular effects either by the activation of ternary complex factor (TCF) [212] or myocardin-related transcription factor (MRTF) dependent gene expression [213]. Besides an established role of SRF and MAPK signaling in cell proliferation, chemerin signaling through CMKLR1 and GPR1 had resulted into different response in different cell lines. It stimulated Rho/MAPK/SRF signaling in human lymphocytes and gastric carcinoma cells, however it exhibited different pattern of downstream signaling in the mentioned cell types [209]. In lymphocytes, the mentioned chemerin-induced expression of SRF upregulated ERG1 and cFOS with an antiproliferative effect at the concentration of 10 nM, however such effect was not observed at the other tested concentrations (1, 30 and 100 nM). Contrarily, chemerin-induced SRF upregulated vinculin instead of ERG1 and cFOS in gastric carcinoma cells with no effect on cell proliferation at all tested concentrations [209].
Since ERG1 inhibits cell proliferation and promotes apoptosis through activating p53 pathway [214,215], chemerin induced upregulation of ERG1 could possibly account for its tumor suppressive properties. However, the role of cFOS and vinculin in cancer differs in different types of malignancies. The upregulation of cFOS by chemerin signaling is reported in a number of cell lines [209,216,217]. Although cFOS was initially believed to act as an oncogene [218,219], its role in cancer progression stands dubious since several studies have reported its cancer suppressive potential in hepatocellular carcinoma [220], ovarian [221,222] and prostate cancer [223]. Likewise, vinculin is an adhesion related protein whose expression is negatively associated with cancer metastasis and poor prognosis [224], its upregulation in gastric cancer patients is positively associated with poor prognosis and shorter survival [225]. As we have mentioned the opposite role of chemerin in human lymphocytes and gastric carcinoma cells with its cancer suppressive effect in the former and cancer supportive potential in the later [209], the chemerin-induced upregulation of cFOS and vinculin could possibly be attributed to the mentioned dual behavior of chemerin in hepatocellular [220,226] and gastric carcinoma [225] respectively. Moreover, chemerin is found to inhibit invasion and metastasis of hepatocellular carcinoma cells by upregulating PTEN and downregulating p-Akt activities [196].
The role of the polarization of naive macrophages to either M1 or M2 phenotype in cancer progression and metastasis has been established already [227]. While M1 macrophages exhibit tumor retardation and impede the progression and metastasis of cancer cells, the M2 phenotype exerts the opposite effect. Because of their anti-inflammatory and pro-tumor attributes, a higher infiltration of cancer tissue with M2 macrophages is reported to be associated with poor prognosis and less survival in different cancers [228–230]. Contrarily, the pro-inflammatory and tumor suppressive properties of M1 macrophages help these macrophages to suppress the invasion and spread of cancer. For instance, a higher infiltration of M1 phenotype in cancerous tissue (M2/M1 < 3) showed better survival with significant inhibition of cancer progression in colon cancer patients [231]. Out of the two mentioned macrophages populations, chemerin exerts a chemoattractant effect on CMKLR1 expressing M1 macrophages selectively [232]. Hence, the chemerin-induced infiltration of M1 macrophages in the cancerous tissue could be attributed as one of the mechanisms by which chemerin exerts its cancer suppressive effect. In addition, the chemerin enhanced infiltration of CMKLR1 positive natural killer cells (NK) in melanoma with a relatively reduced colocalization of plasmacytoid dendtritic cells (pDC) and myeloid-derived suppressor cells resulted into the inhibition of tumorigenesis by bolstering the immune response [233]. The downregulation of CMKLR1 receptors attenuated this tumor-suppressive effect of chemerin, indicating a major contribution of NK cell infiltration in the chemerin induced tumor suppression.
Now we discuss the properties of chemerin that support the invasion and spread of cancer cells. Being a chemoattractant adipokine, chemerin has a strong potential to bolster the obesity associated chronic inflammation [234]. This notion is further attested by the fact that the serum chemerin levels are positively associated with inflammatory cytokines and glycated hemoglobin and both of these factors are associated with chronic inflammation [235,236]. These inflammatory cytokines, particularly IL-6, IL-1β and TNFα upregulate the expression of CMKLR1 receptors on the surface of the endothelial cells [237]. The CMKLR1 signaling was shown to exert angiogenic effect on the endothelial cells via MAPK and PI3K/Akt dependent upregulation of MMP2 and MMP9 expression [237]. Chemerin upregulated the VEGF expression in endothelial cells [238] and gastric cancer cells along with an upregulation of p38 MAP kinase, ERK1/2 and MMP7 [239]. The angiogenic potential of chemerin was also confirmed in the mice model [240]. It was reported to induce proliferation, migration, and tube formation in HUVEC cells through phosphorylation of Akt and p42/44 ERK. Further, the mentioned effects were completely abrogated by silencing CMKLR1 receptors in HUVEC cells that suggested a strong angiogenic potential of chemerin. Apart from its chemoattractant effect on macrophages and NK, it also exerts significant migration and recruitment of pDC through CMKLR1 receptors [241]. Although a further study is required to authenticate such chemerin-induced infiltration of pDC in cancers, it is important to mention here that the tumor microenvironment modifies the tumor infiltrating pDC. The downregulation of IRF7 pathway [242] as well as other factors of tumor microenvironment such as PGE2 and TGFβ [243] abrogate the ability of pDC to release INFα. The lack of INFα results into an immunosuppressive tumor microenvironment that facilitates the progression and metastasis of cancer cells as reported in patients with head and neck cancer [244].
Neutrophil Gelatinase-Associated Lipocalin/Lipocalin 2
Lipocalin 2 (LCN2) is a glycoproteins that performs various physiological functions such as the remodeling of adipose tissue, iron homeostasis, immune responses and the transport of hydrophilic substances across the cell membrane. The levels of LCN2 are positively associated with obesity [245]. Although a few studies have reported its antiproliferative and anti-metastatic role in cancer [246,247], a considerable part of the literature presents an entirely different picture with LCN2 as a promoter of cell proliferation, invasion, metastasis and poor prognosis in different types of cancers (Please see Supplementary Material Table 1). With a 27-fold higher expression in pancreatic cancer cells (compared with normal ductal cells) and an elevated serum level in 94% of the pancreatic cancer patients, LCN2 is reported as an early detection marker for pancreatic malignancies [248]. Likewise, its expression is upregulated in the advanced stages (stage II and III) of breast cancer patients suggesting its critical role in cancer invasion and nodal metastasis [249].
The expression of LCN2 is significantly contributed by the regulatory T cells of the tumor microenvironment. On one side, these regulatory T cells exhibit an immunosuppressive effect that favors the survival of cancer cells within the tumor microenvironment [250]. Additionally, the FoxP3+ regulatory cells release pro-inflammatory cytokines such as IL17, IL1β and TNFα that help recruit NF-κB on the LCN2 promotor region [251]. Hence, the release of these pro-inflammatory cytokines from the regulatory T cells of the tumor microenvironment directly upregulates the expression and levels of LCN2 in the cancer cells. Moreover, the C-X-C motif chemokine receptor 7 signaling in cancer cells upregulates the expression and release of MMP2 and MMP9 into the extracellular matrix [252]. LCN2 treatment has itself resulted into an increased expression of MMP1, MMP3 and MMP9 along with an upregulation of IL-1β, IL-6, IL-8, ICAM-1 and C-X-C motif chemokine 2/monocyte chemoattractant protein 1 (MCP-1) in pancreatic cancer stellate cells [253]. The release of MMP9 and LCN2 into the extracellular space leads to the formation of LCN2-MMP9 complex into the extracellular matrix. In such complex, LCN2 activates MMP9 [254], increases its stability by preventing its degradation [255] and promotes its enzymatic activity [256]. LCN2-MMP9 complex has been found to promote proliferation and metastasis in various cancer cells such as the gastric [255], breast [257] and thyroid carcinoma cells [258]. Higher levels of LCN2-MMP9 complex are found in the urine samples of cancer patients suggesting the significance of the extracellular LCN2-MMP9 complex in the proliferation and metastasis of cancer cells [259]. The role of LCN2 in cancer metastasis is further confirmed by the fact that its deficiency in murine breast cancer cells had resulted into a significant reduction in the size and metastasis of the primary tumors with a reduced MMP9 levels in blood [260]. Likewise, the downregulation of NF-κB in human thyroid carcinoma cells had resulted into a failure in growing the cancer cells and inability to grow the xenograft via downregulating LCN2 [261]. Contrarily, its upregulation has produced opposite effects in human PANC1 pancreatic carcinoma cells with an aggressive invasion and metastasis [262]. LCN2 promotes EMT by upregulating mesenchymal markers such as fibronectin and vimentin in breast cancer cells while inhibiting the epithelial marker E-cadherin [249]. Silencing of LCN2 expression in the highly aggressive breast cancer cell line MDA-MB-231 has resulted into a diminished mesenchymal transition that has further confirmed the involvement of LCN2 in promoting EMT [249].
The upregulation of Ras/MAPK signaling in Ras-transformed cells promotes the phosphorylation and degradation of E-cadherin [263]. LCN2 was reported to block EMT in Ras-transformed 4T1 cells by antagonizing such degradation of E-cadherin [263]. It also exhibited anti-metastatic and antiangiogenic effect in these cells via inhibiting Ras-induced VEGF and upregulating caveolin-1 and anti-angiogenic thrombospondin. Contrarily, it promoted invasion, metastasis and EMT in non Ras-transformed 4T1 cells via attenuating PI3K/Akt pathway [264]. It is important to mention here that the PI3K/Akt signaling promotes cancer cell survival by inhibiting tumor suppressor tuberous sclerosis complex 2 (TSC2). However, it exhibits both suppressive and promoting effects on cancer cell invasion and metastasis. For instance, PI3K/Akt was reported to suppress cancer metastasis in breast cancer cells [265] and promote cell migration and invasion in esophageal [266] and renal carcinoma [267]. The question arises as to how the LCN2-induced attenuation of PI3K/Akt pathway could account for the pro-metastatic effect of LCN2. It is recently reported that the underlying mechanism of Akt-induced metastasis involves the upregulation of MMP2 and MMP9 [267]. As the MMPs are upregulated by LCN2 itself [253] as well as by other players of the tumor microenvironment such as the CXC motif chemokines [252], the LCN2 mediated suppression of Akt could not impede the invasion and metastasis of cancer cells. Further, the downregulation of Akt signaling could promote metastasis via TSC2 mediated activation of Rho/GTPase signaling [268]. A higher expression of TSC2 in breast cancer patients was positively associated with aggressive metastasis and poor survival rate [268]. Since Akt boosts the phosphorylation and degradation of the nuclear factor of activated T-cells (NFAT) through ubiquitin ligase MDM2 [269], the downregulation of Akt signaling could also promote metastasis by enhancing the transcriptional activity of NFAT [269].
Once synthesized within the cancer cells, LCN2 is released into the extracellular matrix where it forms LCN2-catecholate-Fe3+ (LCF) complex. Moreover, the LCF complex enters the cancer cells through the LCN2 receptors, increasing the intracellular iron concentration. It is important to mention here that the hydroxy groups of catechol reduce the iron ions from ferric to ferrous state in the absence of LCN2. This could further precipitate into the generation of ROS that could be detrimental for the survival of the cancer cells. Since LCN2 binds with catechol through O-sulfonation or O-hydroxylation, it prevents the reduction of iron ions into the ferrous state [270]. Thereby it blocks the generation of ROS, a change that prevents the anchorage free cancer cells from the caspase-independent ROS mediated cell death mechanisms [271]. Additionally, the increase in cytosolic iron level inactivates the pro-apoptotic Bcl-2-like protein 11 (BIM) [272]. The increase in intracellular iron stores in breast cancer cells has resulted into a hepcidin mediated degradation of ferroportin, attenuating iron efflux and promoting cancer cell survival and proliferation [273].
Uridine
Uridine is a nucleoside involved in many physiological functions such as the homeostatic regulation of body temperature, glycogen storage and energy balance, however its physiological role has not gained that much attention of the research community as compared to adenosine. It is synthesized in hepatocytes and adipocytes where the adipocytes are the major source of uridine synthesis under fasting conditions [274]. Plasma uridine levels increase during fasting that attenuate body metabolism through hypothalamic pathways, thus reducing the body temperature [274]. Contrarily, a meal induces the secretion of bile into the small intestine, resulting into the biliary clearance of uridine which precipitates into a postprandial decline in plasma uridine levels. Interestingly, such postprandial decrease in plasma uridine levels is not seen in obese individuals. The results from a recent study has shown a postprandial increase in plasma uridine levels in obese individuals [275]. Since the plasma levels of uridine are increased in obesity, we will now focus on the possible role of uridine and its salvage pathway in supporting/promoting cancer cells.
Till date, no specific receptors for uridine have been identified. However, most of its physiological functions are conferred through G protein coupled purine receptors such as P2Y2, P2Y4, P2Y6 and P2Y14. The mentioned purine receptors, especially P2Y2 and P2Y6 play a significant role in tumorigenesis and metastasis [276,277]. UDP released by the doxorubicin treated breast cancer cells, upregulates the expression as well as the enzymatic effect of MMP9 via P2Y6 mediated activation of MAPK and NF-κB signaling [278]. Such UDP-induced effect of MMP9 was found to promote metastasis of breast cancer cells in both the in vitro and in vivo models of breast cancer.
The overexpression of two important enzymes of uridine salvage pathway i.e., uridine phosphorylase and uridine-cytidine kinase in different malignancies such as oral squamous cell carcinoma [279], breast [280], thyroid [281] and lung cancers [282] is considered as an indicator of nodal metastasis and poor prognosis. Guan and coworkers [281] have investigated the involvement of uridine phosphorylase on the proliferation and metastasis of the thyroid carcinoma cells. The inhibition of uridine phosphorylase expression resulted into a significant decline in proliferation, invasion and metastasis of cancer cells. Their findings suggested a possible involvement of uridine phosphorylase in the regulation of EMT, however, a further investigation is required to validate this postulation.
Uric Acid
The biosynthesis of uric acid (UA) from xanthine, during the adenine and guanine degradation, takes place in skeletal muscles, liver, and adipocytes. A number of clinical studies have confirmed a positive correlation between serum UA levels and obesity [283–285]. While evaluating the correlation between cancer and hyperuricemia, one must consider the fact that the higher serum UA levels in cancer patients could be consequent to the cancer itself. The extracellular release of UA that is secondary to the cancer-associated cell necrosis may account for hyperuricemia in cancer patients. However, the findings of several prospective studies have confirmed a significantly higher occurrence of cancer in individuals with higher serum UA levels, proposing hyperuricemia as a potential risk factor for developing cancer [286–288]. A serum UA level above 358 µM was reported as the prospective risk factor in harboring prostate cancers [289]. Moreover, the survival rate of cancer patients with higher serum UA levels is reported as a potential indicator of poor prognosis and diminished survival rate in a number of malignancies such as renal [290], colorectal [291] gastric cancers [292] and osteosarcoma [293]. Now we focus on the possible underlying mechanism that could be attributed to the cancer-supportive nature of UA.
UA exhibits a dual redox behavior with its potential anti-oxidant effect in the extracellular fluid [294] and an oxidant nature in the intracellular environment [295,296]. It activates NADPH mediated oxidative stress in a number of cells such as the adipocytes [297], endothelial cells [298], vascular smooth muscles cells [299], proximal tubular cells [300] and hepatocytes [301]. It is important to mention here that the generation of reactive oxygen species (ROS) during the intracellular oxidative stress is one of the non-apoptotic cell-death mechanisms that has been observed in the extracellular matrix-detached cells [271]. The cancer cells neutralize ROS by upgrading Nrf2 expression to survive and metastasize [302]. By activating NADPH oxidase (NOX) activity, UA induces oxidative stress that might be oppressive for cancer cell survival and metastasis. Moreover, UA was initially hypothesized as a potential anti-cancer agent amid its remarkable extracellular antioxidant properties [303]. However, the extensive research in the following years presented an entirely different picture where UA appeared to be associated with higher cancer risk, poor prognosis and aggressive metastasis as discussed above.
An elevated level of xanthine oxidoreductase (XOR) helps depleting the intracellular xanthine and hypoxanthine levels by converting these metabolites into UA along with ROS generation. A higher expression of XOR is hence believed to suppress tumorigenesis as it boosts the degradation of purines, thus making them unavailable for nucleic acid synthesis. That is why, a downregulation of XOR is reported in a number of aggressive malignancies [304–306]. By downregulating XOR, the cancer cells maintain a pool of purines that are utilized into the replication of genetic material in these rapidly dividing cells. The circulating UA enters the cells through special transporter proteins such as urate transporter 1 (Urat1) and glucose transporter 9 (GLUT9). As the intracellular UA levels rise, the activity of XOR is attenuated. The decline in XOR activity precipitates into a higher intracellular purine level that helps the cancer cells to proliferate at a higher rate. Further, hyperuricemia may downregulate the expression of adiponectin via attenuating peroxisome proliferator-activated receptor gamma (PPARγ) in adipocytes [307]. Being a potential inhibitor of cell proliferation, adiponectin serves as an endogenous anticancer marker and its downregulation by UA may facilitate proliferation and metastasis of cancer cells as discussed above. Moreover, the downregulation of XOR in cancer cells is associated with an increased expression of two important factors, the inhibitor of differentiation 1 (ID1) and cyclooxygenase 2 (COX-2) [308,309]. Such upregulation of ID1 and COX-2 is associated with higher migratory properties, metabolic adaptations for survival in drastic conditions, enhanced tumorigenic angiogenesis and metastasis [309–312]. Hence, the hyperuricemia may potentiate invasiveness and metastasis in cancer cells by increasing the ID1 and COX-2 expression via XOR downregulation. Besides, a higher expression of COX-2 and other inflammatory mediators may turn into a hyperuricemia induced chronic inflammatory condition which further promotes the survival and metastasis of cancer cells [313,314].
Besides the downregulation of XOR, the activation of NOX in cancer cells may also upregulate the expression of COX-2 [315]. Being an activator of NOX, UA may upregulate COX-2 expression by NOX pathway as well. Further, UA has been recently reported to increase the expression of proinflammatory cytokine TNF-α and toll like receptor 4 (TLR4) in macrophages [316]. It is important to mention here that the activated tumor-associated macrophages may promote cancer progression by upregulating COX-2 in cancer cells via IL-1β auto amplification [315]. Such auto amplification of IL-1β is found to involve NLRP3 inflammasome activation in cancer-associated macrophages [317]. Once activated, these NLRP3 inflammasomes may drastically promote cancer angiogenesis, endothelial cell proliferation, and immunosuppressive cell activation via releasing IL-1β and IL-18 [318,319]. Importantly, UA is reported to contribute in hepatic steatosis by activating NLRP3 inflammasomes in hepatocytes [296]. Thenceforth, it can be anticipated that UA may exhibit similar kind of inflammasome activation in cancer-associated immune cells to spur tumor invasion and metastasis. Further experiments are required however to attest such claim.
Conclusion
The correlation between obesity and cancer is well-established. However, the obesity mediators contribute to the tumor growth and metastasis via extensive cell signaling pathways that requires a comprehensive review. A general conclusion of the above discussion is concisely presented in Figure 4. The adipokines regulate the proliferation and metastasis of cancer cells through complicated and interrelated pathways. Despite of their well-documented role in the disease progression, the underlying mechanisms exploited by these adipokines remain largely skeptical. For some of the adipokines, such as Ang 1, chemerin and LCN2, the consideration of their role as an anti-cancer or pro-cancer stands debatable because of their dubious role in activating both the cancer suppressive and cancer promoting signaling (please see the supplementary material). Moreover, a given adipokine may provoke proliferation and metastasis in one type of disease and may impede cancer progression in another type of cancer [209]. A number of adipokines potentiate the belligerence of the disease by promoting cell proliferation, metastasis and abating the immune checkpoints. These cytokines include leptin, resistin and ANGLPs. Contrarily, omentin and adiponectin exhibit an inhibitory effect on cancer cells by impeding cell proliferation, metastasis and inducing apoptosis. Such protective role of these anti-cancer adipokines is attested by the fact that the expression of these mediators is downregulated in aggressive cancers. Most of the adipokines promote the spread of cancer cells via upregulating the angiogenic as well as the EMT markers. Some of these markers exhibit strong immunosuppressive effect that helps the cancer cells to escape the immune checkpoints. It is important to mention that the scarcity of either of the three major external factors i.e., the nutrients, growth factors and growing space stops the entry of a cell into the cell cycle at the late G1 phase. This is the major regulatory check point that keeps the normal cells in quiescence under the unfavorable drastic conditions. Contrarily, the cancer cells escape this checkpoint to survive under the extremely harsh conditions. The adipokines provoke such tumorigenic transition by upregulating the expression of Myc, hTERT, E2F1, CycD and other proliferative genes. The adipokine mediated tumorigenic shift of the cellular microenvironment greatly affects the survival rate, remission and therapeutic success. Since the adipokine levels greatly vary among individuals, a consideration of patient adipokine profile should not be ignored while predicting the overall disease progression.
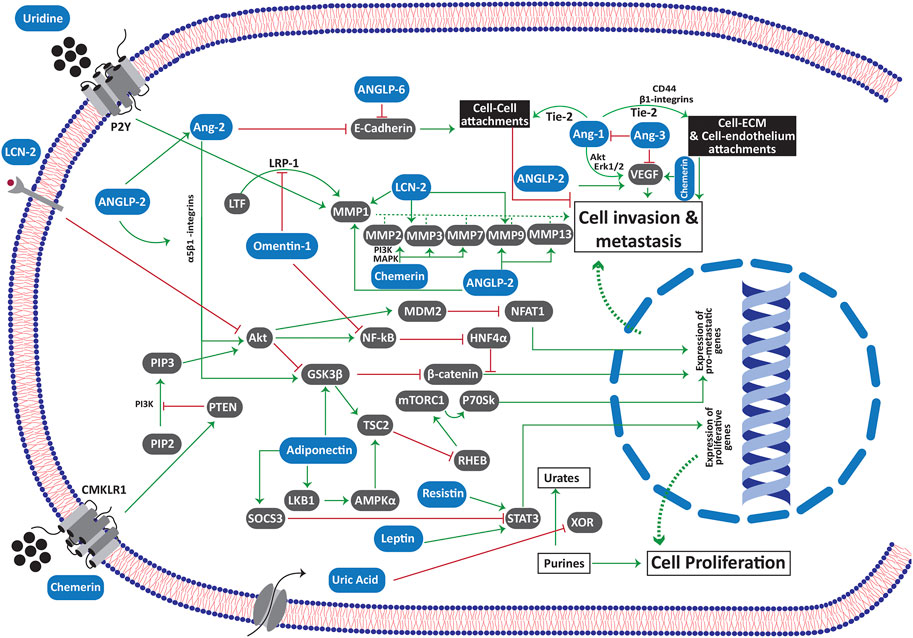
FIGURE 4. Mediation of different cell signaling pathways by adipokines in cancer cell. Abbreviations: Akt protein kinase B; ANG angiopoietin; ANGPL angiopoietin like protein; CMKLR chemerin like receptor; ERK extracellular-signal-related kinase; GSK3 glycogen synthase kinase 3; HNF hepatic nuclear factor; LCN2 lipocalin 2; LKB liver kinase B; LRP low intensity lipoprotein receptor related protein; :LTF lactotransferrin; MDM2 mouse double minute 2 homolog; MMP matrix metalloproteinase; NFAT nuclear factor of activated T cells; NF-κB nuclear factor kappa B; PIP2 phosphatidylinositol 4,5-bisphosphate; PIP3 phosphatidylinositol 3,4,5-triphosphate; PTEN phosphatase and tensin homolog; RHEB Ras homolog enriched in brain; SOCS3 suppressor of cytokine signaling 3; STAT3 signal transducer and activator of transcription 3; TSC tuberous sclerosis complex; VEGF vascular endothelial growth factor; XOR xanthine oxidoreductase.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was supported by Research on Identification and Quality Evaluation of Cross-border Traditional Medicine Resources between China and Pakistan 2020E01011, Wei Hongyan, Xinjiang Institute of Traditional Chinese Medicine and Ethnic Medicine.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the higher education commission (HEC) of Pakistan for providing full access to the HEC digital library and the international research database. Authors thank Ajman university for supporting a part of APC.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.por-journal.com/articles/10.3389/pore.2021.1609828/full#supplementary-material
References
1. Madsen, MS, Siersbæk, R, Boergesen, M, Nielsen, R, and Mandrup, S. Peroxisome Proliferator-Activated Receptor γ and C/EBPα Synergistically Activate Key Metabolic Adipocyte Genes by Assisted Loading. Mol Cel Biol (2014) 34(6):939–54. doi:10.1128/mcb.01344-13
2. Shamsi, F, Xue, R, Huang, TL, Lundh, M, Liu, Y, Leiria, L, et al. FGF6 and FGF9 Regulate UCP1 Expression Independent of Brown Adipogenesis. Nat Commun (2020) 11(1):1421. doi:10.1038/s41467-020-15055-9
3. Leitner, BP, Huang, S, Brychta, RJ, Duckworth, CJ, Baskin, AS, McGehee, S, et al. Mapping of Human Brown Adipose Tissue in Lean and Obese Young Men. Proc Natl Acad Sci USA (2017) 114(32):8649–54. doi:10.1073/pnas.1705287114
4. Wu, J, Boström, P, Sparks, LM, Ye, L, Choi, JH, Giang, A-H, et al. Beige Adipocytes Are a Distinct Type of Thermogenic Fat Cell in Mouse and Human. Cell (2012) 150(2):366–76. doi:10.1016/j.cell.2012.05.016
5. Garcia, RA, Roemmich, JN, and Claycombe, KJ. Evaluation of Markers of Beige Adipocytes in white Adipose Tissue of the Mouse. Nutr Metab (2016) 13:24. doi:10.1186/s12986-016-0081-2
6. Okla, M, Ha, J-H, Temel, RE, and Chung, S. BMP7 Drives Human Adipogenic Stem Cells into Metabolically Active Beige Adipocytes. Lipids (2015) 50(2):111–20. doi:10.1007/s11745-014-3981-9
7. Liu, P, Huang, S, Ling, S, Xu, S, Wang, F, Zhang, W, et al. Foxp1 Controls Brown/beige Adipocyte Differentiation and Thermogenesis through Regulating β3-AR Desensitization. Nat Commun (2019) 10(1):5070. doi:10.1038/s41467-019-12988-8
8. Organization, WH. Obesity And Overweight. News Room: Fact Sheets 2021. Available from: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight.
9. Reeves, GK, Pirie, K, Beral, V, Green, J, Spencer, E, and Bull, D. Cancer Incidence and Mortality in Relation to Body Mass index in the Million Women Study: Cohort Study. Bmj (2007) 335(7630):1134. doi:10.1136/bmj.39367.495995.ae
10. Arnold, M, Pandeya, N, Byrnes, G, Renehan, AG, Stevens, GA, Ezzati, M, et al. Global burden of Cancer Attributable to High Body-Mass index in 2012: a Population-Based Study. Lancet Oncol (2015) 16(1):36–46. doi:10.1016/s1470-2045(14)71123-4
11. Odutola, MK, Olukomogbon, T, Igbinoba, F, Otu, TI, Ezeome, E, Hassan, R, et al. Cancers Attributable to Overweight and Obesity from 2012 to 2014 in Nigeria: A Population-Based Cancer Registry Study. Front Oncol (2019) 9:460. doi:10.3389/fonc.2019.00460
12. de Gonzalez, AB, Sweetland, S, and Spencer, E. A Meta-Analysis of Obesity and the Risk of Pancreatic Cancer. Br J Cancer (2003) 89(3):519–23. doi:10.1038/sj.bjc.6601140
13. Holt, DJ, and Grainger, DW. Senescence and Quiescence Induced Compromised Function in Cultured Macrophages. Biomaterials (2012) 33(30):7497–507. doi:10.1016/j.biomaterials.2012.06.099
14. Liu, H, Adler, AS, Segal, E, and Chang, HY. A Transcriptional Program Mediating Entry into Cellular Quiescence. Plos Genet (2007) 3(6):e91. doi:10.1371/journal.pgen.0030091
15. Seyfried, TN, and Huysentruyt, LC. On the Origin of Cancer Metastasis. Crit Rev oncogenesis (2013) 18(1-2):43–73. doi:10.1615/critrevoncog.v18.i1-2.40
16. Piva de Freitas, P, Senna, CG, Tabai, M, Chone, CT, and Altemani, A. Metastatic Basal Cell Carcinoma: A Rare Manifestation of a Common Disease. Case Rep Med (2017) 2017:8929745. doi:10.1155/2017/8929745
17. García Franco, CE, Torre, W, Tamura, A, Guillén-Grima, F, San-Julian, M, Martin-Algarra, S, et al. Long-term Results after Resection for Bone Sarcoma Pulmonary Metastases. Eur J Cardiothorac Surg (2010) 37(5):1205–8. doi:10.1016/j.ejcts.2009.11.026
18. Cho, YK, Lee, J, Kim, HS, Park, JY, Lee, WJ, Kim, YJ, et al. Metabolic Health Is a Determining Factor for Incident Colorectal Cancer in the Obese Population: A Nationwide Population‐based Cohort Study. Cancer Med (2021) 10(1):220–9. doi:10.1002/cam4.3607
19. James, DB, Mary, KG, and Christian, W. TNM Classification of Malignant Tumours. Wiley-Blackwell (2016).
20. McAlpine, J, Leon-Castillo, A, and Bosse, T. The Rise of a Novel Classification System for Endometrial Carcinoma; Integration of Molecular Subclasses. J Pathol (2018) 244(5):538–49. doi:10.1002/path.5034
21. Yanovich, G, Agmon, H, Harel, M, Sonnenblick, A, Peretz, T, and Geiger, T. Clinical Proteomics of Breast Cancer Reveals a Novel Layer of Breast Cancer Classification. Cancer Res (2018) 78(20):6001–10. doi:10.1158/0008-5472.CAN-18-1079
22. Ilie, M, Hofman, V, Dietel, M, Soria, J-C, and Hofman, P. Assessment of the PD-L1 Status by Immunohistochemistry: Challenges and Perspectives for Therapeutic Strategies in Lung Cancer Patients. Virchows Arch (2016) 468(5):511–25. doi:10.1007/s00428-016-1910-4
23. Mamatjan, Y, Agnihotri, S, Goldenberg, A, Tonge, P, Mansouri, S, Zadeh, G, et al. Molecular Signatures for Tumor Classification. J Mol Diagn (2017) 19(6):881–91. doi:10.1016/j.jmoldx.2017.07.008
24. Caldefie-Chézet, F, Damez, M, de Latour, M, Konska, G, Mishellani, F, Fusillier, C, et al. Leptin: A Proliferative Factor for Breast Cancer? Biochem Biophysical Res Commun (2005) 334(3):737–41. doi:10.1016/j.bbrc.2005.06.077
25. Yang, G, Fan, W, Luo, B, Xu, Z, Wang, P, Tang, S, et al. Circulating Resistin Levels and Risk of Colorectal Cancer: A Meta-Analysis. Biomed Res Int (2016) 2016:7367485. doi:10.1155/2016/7367485
26. Hu, W-W, Tang, C-H, Sun, Y, Lu, T-T, Jiang, P, Wu, Y-M, et al. Correlation between Resistin Gene Polymorphism and Clinical Aspects of Lung Cancer. Medicine (2017) 96(52):e9485. doi:10.1097/md.0000000000009485
27. Diakowska, D, Markocka-Mączka, K, Szelachowski, P, and Grabowski, K. Serum Levels of Resistin, Adiponectin, and Apelin in Gastroesophageal Cancer Patients. Dis Markers (2014) 2014:619649. doi:10.1155/2014/619649
28. Cymbaluk-Płoska, A, Chudecka-Głaz, A, Pius-Sadowska, E, Sompolska-Rzechuła, A, Machaliński, B, and Menkiszak, J. Circulating Serum Level of Visfatin in Patients with Endometrial Cancer. Biomed Res Int (2018) 2018:8576179. doi:10.1155/2018/8576179
29. Vallega, KA, Liu, N, Myers, JS, Yu, K, and Sang, Q-XA. Elevated Resistin Gene Expression in African American Estrogen and Progesterone Receptor Negative Breast Cancer. PloS one (2016) 11(6):e0157741. doi:10.1371/journal.pone.0157741
30. Funcke, J-B, and Scherer, PE. Beyond Adiponectin and Leptin: Adipose Tissue-Derived Mediators of Inter-organ Communication. J Lipid Res (2019) 60–16481684. doi:10.1194/jlr.R094060
31. Wang, H, Liang, X, Li, M, Tao, X, Tai, S, Fan, Z, et al. Chemokine (CCmotif) Ligand 18 Upregulates Slug Expression to Promote Stem‐cell like Features by Activating the Mammalian Target of Rapamycin Pathway in Oral Squamous Cell Carcinoma. Cancer Sci (2017) 108(8):1584–93. doi:10.1111/cas.13289
32. Park, J, and Scherer, PE. Leptin and Cancer: from Cancer Stem Cells to Metastasis. Endocrine-related cancer (2011) 18(4):C25–C29. doi:10.1530/erc-11-0163
33. Hardwick, JCH, Van Den Brink, GR, Offerhaus, GJ, Van Deventer, SJH, and Peppelenbosch, MP. Leptin Is a Growth Factor for Colonic Epithelial Cells. Gastroenterology (2001) 121(1):79–90. doi:10.1053/gast.2001.25490
34. Attoub, S, Noe, V, Pirola, L, Bruyneel, E, Chastre, E, Mareel, M, et al. Leptin Promotes Invasiveness of Kidney and Colonic Epithelial Cells via Phosphoinositide 3‐kinase‐, Rho‐, and Rac‐dependent Signaling Pathways. FASEB j (2000) 14(14):2329–38. doi:10.1096/fj.00-0162
35. Buyse, M, Berlioz, F, Guilmeau, S, Tsocas, A, Voisin, T, Péranzi, G, et al. PepT1-mediated Epithelial Transport of Dipeptides and Cephalexin Is Enhanced by Luminal Leptin in the Small Intestine. J Clin Invest (2001) 108(10):1483–94. doi:10.1172/jci13219
36. Koda, M, Sulkowska, M, Kanczuga-Koda, L, Surmacz, E, and Sulkowski, S. Overexpression of the Obesity Hormone Leptin in Human Colorectal Cancer. J Clin Pathol (2007) 60(8):902–6. doi:10.1136/jcp.2006.041004
37. Gu, L, Wang, C-D, Cao, C, Cai, L-R, Li, D-H, and Zheng, Y-Z. Association of Serum Leptin with Breast Cancer: A Meta-Analysis. Medicine (2019) 98(5):e14094. doi:10.1097/md.0000000000014094
38. Jeong, Y-J, Bong, J-G, Park, S-H, Choi, J-H, and Oh, H-K. Expression of Leptin, Leptin Receptor, Adiponectin, and Adiponectin Receptor in Ductal CarcinomaIn Situand Invasive Breast Cancer. J Breast Cancer (2011) 14(2):96–103. doi:10.4048/jbc.2011.14.2.96
39. Harris, HR, Tworoger, SS, Hankinson, SE, Rosner, BA, and Michels, KB. Plasma Leptin Levels and Risk of Breast Cancer in Premenopausal Women. Cancer Prev Res (2011) 4(9):1449–56. doi:10.1158/1940-6207.capr-11-0125
40. Gu, F, Kraft, P, Rice, M, and Michels, KB. Leptin and Leptin Receptor Genes in Relation to Premenopausal Breast Cancer Incidence and Grade in Caucasian Women. Breast Cancer Res Treat (2012) 131(1):17–25. doi:10.1007/s10549-011-1778-6
41. Ferla, R, Bonomi, M, Otvos, L, and Surmacz, E. Glioblastoma-derived Leptin Induces Tube Formation and Growth of Endothelial Cells: Comparison with VEGF Effects. BMC cancer (2011) 11:303. doi:10.1186/1471-2407-11-303
42. Wang, X-J, Yuan, S-L, Lu, Q, Lu, Y-R, Zhang, J, Liu, Y, et al. Potential Involvement of Leptin in Carcinogenesis of Hepatocellular Carcinoma. Wjg (2004) 10(17):2478–81. doi:10.3748/wjg.v10.i17.2478
43. Zhou, J, Lei, W, Shen, L, Luo, H-S, and Shen, Z-X. Primary Study of Leptin and Human Hepatocellular Carcinoma In Vitro. Wjg (2008) 14(18):2900–4. doi:10.3748/wjg.14.2900
44. Saxena, NK, Sharma, D, Ding, X, Lin, S, Marra, F, Merlin, D, et al. Concomitant Activation of the JAK/STAT, PI3K/AKT, and ERK Signaling Is Involved in Leptin-Mediated Promotion of Invasion and Migration of Hepatocellular Carcinoma Cells. Cancer Res (2007) 67(6):2497–507. doi:10.1158/0008-5472.can-06-3075
45. Ptak, A, Kolaczkowska, E, and Gregoraszczuk, EL. Leptin Stimulation of Cell Cycle and Inhibition of Apoptosis Gene and Protein Expression in OVCAR-3 Ovarian Cancer Cells. Endocrine (2013) 43(2):394–403. doi:10.1007/s12020-012-9788-7
46. Pan, W, Allison, MB, Sabatini, P, Rupp, A, Adams, J, Patterson, C, et al. Transcriptional and Physiological Roles for STAT Proteins in Leptin Action. Mol Metab (2019) 22:121–31. doi:10.1016/j.molmet.2019.01.007
47. Saxena, NK, Vertino, PM, Anania, FA, and Sharma, D. Leptin-induced Growth Stimulation of Breast Cancer Cells Involves Recruitment of Histone Acetyltransferases and Mediator Complex to CYCLIN D1 Promoter via Activation of Stat3. J Biol Chem (2007) 282(18):13316–25. doi:10.1074/jbc.m609798200
48. Saxena, NK, Titus, MA, Ding, X, Floyd, J, Srinivasan, S, Sitaraman, SV, et al. Leptin as a Novel Profibrogenic Cytokine in Hepatic Stellate Cells: Mitogenesis and Inhibition of Apoptosis Mediated by Extracellular Regulated Kinase (Erk) and Akt Phosphorylation. FASEB j (2004) 18(13):1612–4. doi:10.1096/fj.04-1847fje
49. Higurashi, T, Endo, H, Uchiyama, T, Uchiyama, S, Yamada, E, Ohkubo, H, et al. Conditional Knockout of the Leptin Receptor in the Colonic Epithelium Revealed the Local Effects of Leptin Receptor Signaling in the Progression of Colonic Tumors in Mice. Carcin (2014) 35(9):2134–41. doi:10.1093/carcin/bgu135
50. Xiong, H, Zhang, Z-G, Tian, X-Q, Sun, D-F, Liang, Q-C, Zhang, Y-J, et al. Inhibition of JAK1, 2/STAT3 Signaling Induces Apoptosis, Cell Cycle Arrest, and Reduces Tumor Cell Invasion in Colorectal Cancer Cells. Neoplasia (2008) 10(3):287–97. doi:10.1593/neo.07971
51. Lin, Q, Lai, R, Chirieac, LR, Li, C, Thomazy, VA, Grammatikakis, I, et al. Constitutive Activation of JAK3/STAT3 in Colon Carcinoma Tumors and Cell Lines. Am J Pathol (2005) 167(4):969–80. doi:10.1016/s0002-9440(10)61187-x
52. Nowakowska-Zajdel, E, Mazurek, U, Stachowicz, M, Niedworok, E, Fatyga, E, and Muc-Wierzgoń, M. Cellular Signal Transduction Pathways by Leptin in Colorectal Cancer Tissue: Preliminary Results. ISRN Endocrinol (2011) 2011:575397. doi:10.5402/2011/575397
53. Mittenbühler, MJ, Sprenger, H-G, Gruber, S, Wunderlich, CM, Kern, L, Brüning, JC, et al. Hepatic Leptin Receptor Expression Can Partially Compensate for IL-6Rα Deficiency in DEN-Induced Hepatocellular Carcinoma. Mol Metab (2018) 17:122–33. doi:10.1016/j.molmet.2018.08.010
54. Zheng, Q, Banaszak, L, Fracci, S, Basali, D, Dunlap, SM, Hursting, SD, et al. Leptin Receptor Maintains Cancer Stem-like Properties in Triple Negative Breast Cancer Cells. Endocrine-related cancer (2013) 20(6):797–808. doi:10.1530/erc-13-0329
55. Thiagarajan, PS, Zheng, Q, Bhagrath, M, Mulkearns-Hubert, EE, Myers, MG, Lathia, JD, et al. STAT3 Activation by Leptin Receptor Is Essential for TNBC Stem Cell Maintenance. Endocr Relat Cancer (2017) 24(8):415–26. doi:10.1530/erc-16-0349
56. Aparicio, T, Kotelevets, L, Tsocas, A, Laigneau, JP, Sobhani, I, Chastre, E, et al. Leptin Stimulates the Proliferation of Human colon Cancer Cells In Vitro but Does Not Promote the Growth of colon Cancer Xenografts in Nude Mice or Intestinal Tumorigenesis in ApcMin/+ Mice. Gut (2005) 54(8):1136–45. doi:10.1136/gut.2004.060533
57. Deo, DD, Rao, AP, Bose, SS, Ouhtit, A, Baliga, SB, Rao, SA, et al. Differential Effects of Leptin on the Invasive Potential of Androgen-dependent and -independent Prostate Carcinoma Cells. J Biomed Biotechnol (2008) 2008:163902. doi:10.1155/2008/163902
58. Naviglio, S, Di Gesto, D, Sorrentino, A, Illiano, F, Sorvillo, L, Abbruzzese, A, et al. Leptin Enhances Growth Inhibition by cAMP Elevating Agents through Apoptosis of MDA-MB-231 Breast Cancer Cells. Cancer Biol Ther (2009) 8(12):1183–90. doi:10.4161/cbt.8.12.8562
59. Spina, A, Di Maiolo, F, Esposito, A, Sapio, L, Chiosi, E, Sorvillo, L, et al. cAMP Elevation Down-Regulates β3 Integrin and Focal Adhesion Kinase and Inhibits Leptin-Induced Migration of MDA-MB-231 Breast Cancer Cells. BioResearch open access (2012) 1(6):324–32. doi:10.1089/biores.2012.0270
60. Catalano, S, Mauro, L, Bonofiglio, D, Pellegrino, M, Qi, H, Rizza, P, et al. In Vivo and In Vitro Evidence that PPARγ Ligands Are Antagonists of Leptin Signaling in Breast Cancer. Am J Pathol (2011) 179(2):1030–40. doi:10.1016/j.ajpath.2011.04.026
61. Alshaker, H, Krell, J, Frampton, AE, Waxman, J, Blyuss, O, Zaikin, A, et al. Leptin Induces Upregulation of Sphingosine Kinase 1 in Oestrogen Receptor-Negative Breast Cancer via Src Family Kinase-Mediated, Janus Kinase 2-independent Pathway. Breast Cancer Res : BCR (2014) 16(5):426. doi:10.1186/s13058-014-0426-6
62. Bjørbaek, C, El-Haschimi, K, Frantz, JD, and Flier, JS. The Role of SOCS-3 in Leptin Signaling and Leptin Resistance. J Biol Chem (1999) 274(42):30059–65. doi:10.1074/jbc.274.42.30059
63. Wan, J, Che, Y, Kang, N, and Wu, W. SOCS3 Blocks HIF-1α Expression to Inhibit Proliferation and Angiogenesis of Human Small Cell Lung Cancer by Downregulating Activation of Akt, but Not STAT3. Mol Med Rep (2015) 12(1):83–92. doi:10.3892/mmr.2015.3368
64. Knobelspies, H, Zeidler, J, Hekerman, P, Bamberg-Lemper, S, and Becker, W. Mechanism of Attenuation of Leptin Signaling under Chronic Ligand Stimulation. BMC Biochem (2010) 11:2. doi:10.1186/1471-2091-11-2
65. Bode, J, Ludwig, S, Freitas, CA, Schaper, F, Wammers, M, Melmed, S, et al. The MKK6/p38 Mitogen-Activated Protein Kinase Pathway Is Capable of Inducing SOCS3 Gene Expression and Inhibits IL-6-Induced Transcription. Biol Chem (2001) 382:1447–53. doi:10.1515/bc.2001.178
66. Thompson, KJ, Lau, KN, Johnson, S, Martinie, JB, Iannitti, DA, McKillop, IH, et al. Leptin Inhibits Hepatocellular Carcinoma Proliferation via P38-MAPK-dependent Signalling. Hpb (2011) 13(4):225–33. doi:10.1111/j.1477-2574.2010.00259.x
67. Inagaki-Ohara, K, Mayuzumi, H, Kato, S, Minokoshi, Y, Otsubo, T, Kawamura, YI, et al. Enhancement of Leptin Receptor Signaling by SOCS3 Deficiency Induces Development of Gastric Tumors in Mice. Oncogene (2014) 33(1):74–84. doi:10.1038/onc.2012.540
68. Ramani, K, Yang, H, Xia, M, Ara, AI, Mato, JM, and Lu, SC. Leptin's Mitogenic Effect in Human Liver Cancer Cells Requires Induction of Both Methionine Adenosyltransferase 2A and 2beta. Hepatology (2008) 47(2):521–31. doi:10.1002/hep.22064
69. Stefanou, N, Papanikolaou, V, Furukawa, Y, Nakamura, Y, and Tsezou, A. Leptin as a Critical Regulator of Hepatocellular Carcinoma Development through Modulation of Human Telomerase Reverse Transcriptase. BMC cancer (2010) 10:442. doi:10.1186/1471-2407-10-442
70. Kasiappan, R, Sun, Y, Lungchukiet, P, Quarni, W, Zhang, X, and Bai, W. Vitamin D Suppresses Leptin Stimulation of Cancer Growth through microRNA. Cancer Res (2014) 74(21):6194–204. doi:10.1158/0008-5472.can-14-1702
71. Fukada, T, Ohtani, T, Yoshida, Y, Shirogane, T, Nishida, K, Nakajima, K, et al. STAT3 Orchestrates Contradictory Signals in Cytokine-Induced G1 to S Cell-Cycle Transition. EMBO J (1998) 17(22):6670–7. doi:10.1093/emboj/17.22.6670
72. Harbuzariu, A, Rampoldi, A, Daley-Brown, DS, Candelaria, P, Harmon, TL, Lipsey, CC, et al. Leptin-Notch Signaling axis Is Involved in Pancreatic Cancer Progression. Oncotarget (2017) 8(5):7740–52. doi:10.18632/oncotarget.13946
73. Feldheim, J, Kessler, AF, Schmitt, D, Salvador, E, Monoranu, CM, Feldheim, JJ, et al. Ribosomal Protein S27/Metallopanstimulin-1 (RPS27) in Glioma-A New Disease Biomarker? Cancers (2020) 12(5):1085. doi:10.3390/cancers12051085
74. Cao, D, Luo, Y, Qin, S, Yu, M, Mu, Y, Ye, G, et al. Metallopanstimulin-1 (MPS-1) Mediates the Promotion Effect of Leptin on Colorectal Cancer through Activation of JNK/c-Jun Signaling Pathway. Cel Death Dis (2019) 10(9):655. doi:10.1038/s41419-019-1911-8
75. Yang, Z-Y, Jiang, H, Qu, Y, Wei, M, Yan, M, Zhu, Z-G, et al. Metallopanstimulin-1 Regulates Invasion and Migration of Gastric Cancer Cells Partially through Integrin β4. Carcinogenesis (2013) 34(12):2851–60. doi:10.1093/carcin/bgt226
76. Christopoulos, PF, Msaouel, P, and Koutsilieris, M. The Role of the Insulin-like Growth Factor-1 System in Breast Cancer. Mol Cancer (2015) 14:43. doi:10.1186/s12943-015-0291-7
77. Min, DY, Jung, E, Kim, J, Lee, YH, and Shin, SY. Leptin Stimulates IGF-1 Transcription by Activating AP-1 in Human Breast Cancer Cells. BMB Rep (2019) 52(6):385–90. doi:10.5483/bmbrep.2019.52.6.189
78. Yang, W-H, Chang, A-C, Wang, S-W, Wang, S-J, Chang, Y-S, Chang, T-M, et al. Leptin Promotes VEGF-C Production and Induces Lymphangiogenesis by Suppressing miR-27b in Human Chondrosarcoma Cells. Scientific Rep (2016) 6(1):28647. doi:10.1038/srep28647
79. Carino, C, Olawaiye, AB, Cherfils, S, Serikawa, T, Lynch, MP, Rueda, BR, et al. Leptin Regulation of Proangiogenic Molecules in Benign and Cancerous Endometrial Cells. Int J Cancer (2008) 123(12):2782–90. doi:10.1002/ijc.23887
80. Zhou, W, Guo, S, and Gonzalez-Perez, RR. Leptin Pro-angiogenic Signature in Breast Cancer Is Linked to IL-1 Signalling. Br J Cancer (2011) 104(1):128–37. doi:10.1038/sj.bjc.6606013
81. Gonzalez-Perez, RR, Xu, Y, Guo, S, Watters, A, Zhou, W, and Leibovich, SJ. Leptin Upregulates VEGF in Breast Cancer via Canonic and Non-canonical Signalling Pathways and NFκB/HIF-1α Activation. Cell Signal (2010) 22(9):1350–62. doi:10.1016/j.cellsig.2010.05.003
82. Guo, S, and Gonzalez-Perez, RR. Notch, IL-1 and Leptin Crosstalk Outcome (NILCO) Is Critical for Leptin-Induced Proliferation, Migration and VEGF/VEGFR-2 Expression in Breast Cancer. PloS one (2011) 6(6):e21467. doi:10.1371/journal.pone.0021467
83. Zhao, X, Dong, Y, Zhang, J, Li, D, Hu, G, Yao, J, et al. Leptin Changes Differentiation Fate and Induces Senescence in Chondrogenic Progenitor Cells. Cell Death Dis (2016) 7(4):e2188. doi:10.1038/cddis.2016.68
84. He, J-Y, Wei, X-H, Li, S-J, Liu, Y, Hu, H-L, Li, Z-Z, et al. Adipocyte-derived IL-6 and Leptin Promote Breast Cancer Metastasis via Upregulation of Lysyl Hydroxylase-2 Expression. Cell Commun signaling : CCS (2018) 16(1):100. doi:10.1186/s12964-018-0309-z
85. Jaffe, T, and Schwartz, B. Leptin Promotes Motility and Invasiveness in Human colon Cancer Cells by Activating Multiple Signal-Transduction Pathways. Int J Cancer (2008) 123(11):2543–56. doi:10.1002/ijc.23821
86. Strong, AL, Strong, TA, Rhodes, LV, Semon, JA, Zhang, X, Shi, Z, et al. Obesity Associated Alterations in the Biology of Adipose Stem Cells Mediate Enhanced Tumorigenesis by Estrogen Dependent Pathways. Breast Cancer Res (2013) 15(5):R102. doi:10.1186/bcr3569
87. Yan, D, Avtanski, D, Saxena, NK, and Sharma, D. Leptin-induced Epithelial-Mesenchymal Transition in Breast Cancer Cells Requires β-Catenin Activation via Akt/GSK3- and MTA1/Wnt1 Protein-dependent Pathways. J Biol Chem (2012) 287(11):8598–612. doi:10.1074/jbc.m111.322800
88. Liang, X, Wang, S, Wang, X, Zhang, L, Zhao, H, and Zhang, L. Leptin Promotes the Growth of Breast Cancer by Upregulating the Wnt/β-Catenin Pathway. Exp Ther Med (2018) 16(2):767–71. doi:10.3892/etm.2018.6212
89. Haque, I, Ghosh, A, Acup, S, Banerjee, S, Dhar, K, Ray, A, et al. Leptin-induced ER-α-Positive Breast Cancer Cell Viability and Migration Is Mediated by Suppressing CCN5-Signaling via Activating JAK/AKT/STAT-pathway. BMC cancer (2018) 18(1):99. doi:10.1186/s12885-018-3993-6
90. Knight, BB, Oprea-Ilies, GM, Nagalingam, A, Yang, L, Cohen, C, Saxena, NK, et al. Survivin Upregulation, Dependent on Leptin-EGFR-Notch1 axis, Is Essential for Leptin-Induced Migration of Breast Carcinoma Cells. Endocrine-related cancer (2011) 18(4):413–28. doi:10.1530/erc-11-0075
91. Battle, M, Gillespie, C, Quarshie, A, Lanier, V, Harmon, T, Wilson, K, et al. Obesity Induced a Leptin-Notch Signaling axis in Breast Cancer. Int J Cancer (2014) 134(7):1605–16. doi:10.1002/ijc.28496
92. Mociño-Rodríguez, MD, Santillán-Benítez, JG, Dozal-Domínguez, DS, Hernández-Navarro, MD, Flores-Merino, MV, Sandoval-Cabrera, A, et al. Expression of AdipoR1 and AdipoR2 Receptors as Leptin-Breast Cancer Regulation Mechanisms. Dis Markers (2017) 2017:4862016. doi:10.1155/2017/4862016
93. Wu, D-C, Zhang, M-F, Su, S-G, Fang, H-Y, Wang, X-H, He, D, et al. HEY2, a Target of miR-137, Indicates Poor Outcomes and Promotes Cell Proliferation and Migration in Hepatocellular Carcinoma. Oncotarget (2016) 7(25):38052–63. doi:10.18632/oncotarget.9343
94. Tanaka, T, Terai, Y, Kogata, Y, Ashihara, K, Maeda, K, Fujiwara, S, et al. CD24 Expression as a Marker for Predicting Clinical Outcome and Invasive Activity in Uterine Cervical Cancer. Oncol Rep (2015) 34(5):2282–8. doi:10.3892/or.2015.4257
95. Xiaoping, L, Xiaowei, Z, Leizhen, Z, and Weijian, G. Expression and Significance of CD44 and P-AKT in Pancreatic Head Cancer. World J Surg Oncol (2015) 13:334. doi:10.1186/s12957-015-0746-8
96. Tsai, CF, Chen, JH, Wu, CT, Chang, PC, Wang, SL, and Yeh, WL. Induction of Osteoclast-like Cell Formation by Leptin-Induced Soluble Intercellular Adhesion Molecule Secreted from Cancer Cells. Ther Adv Med Oncol (2019) 11:1758835919846806. doi:10.1177/1758835919846806
97. Dong, Z, Fu, S, Xu, X, Yang, Y, Du, L, Li, W, et al. Leptin-mediated Regulation of ICAM-1 Is Rho/ROCK Dependent and Enhances Gastric Cancer Cell Migration. Br J Cancer (2014) 110(7):1801–10. doi:10.1038/bjc.2014.70
98. Strong, AL, Ohlstein, JF, Biagas, BA, Rhodes, LV, Pei, DT, Tucker, HA, et al. Leptin Produced by Obese Adipose Stromal/stem Cells Enhances Proliferation and Metastasis of Estrogen Receptor Positive Breast Cancers. Breast Cancer Res (2015) 17(1):112. doi:10.1186/s13058-015-0622-z
99. Sabol, RA, Bowles, AC, Côté, A, Wise, R, O'Donnell, B, Matossian, MD, et al. Leptin Produced by Obesity-Altered Adipose Stem Cells Promotes Metastasis but Not Tumorigenesis of Triple-Negative Breast Cancer in Orthotopic Xenograft and Patient-Derived Xenograft Models. Breast Cancer Res : BCR (2019) 21(1):67. doi:10.1186/s13058-019-1153-9
100. Wei, L, Li, K, Pang, X, Guo, B, Su, M, Huang, Y, et al. Leptin Promotes Epithelial-Mesenchymal Transition of Breast Cancer via the Upregulation of Pyruvate Kinase M2. J Exp Clin Cancer Res : CR (2016) 35(1):166. doi:10.1186/s13046-016-0446-4
101. Wang, L, Tang, C, Cao, H, Li, K, Pang, X, Zhong, L, et al. Activation of IL-8 via PI3K/Akt-dependent Pathway Is Involved in Leptin-Mediated Epithelial-Mesenchymal Transition in Human Breast Cancer Cells. Cancer Biol Ther (2015) 16(8):1220–30. doi:10.1080/15384047.2015.1056409
102. Xu, M, Cao, FL, Li, N, Gao, X, Su, X, and Jiang, X. Leptin Induces Epithelial-To-Mesenchymal Transition via Activation of the ERK Signaling Pathway in Lung Cancer Cells. Oncol Lett (2018) 16(4):4782–8. doi:10.3892/ol.2018.9230
103. Fazolini, NP, Cruz, AL, Werneck, MB, Viola, JP, Maya-Monteiro, CM, and Bozza, PT. Leptin Activation of mTOR Pathway in Intestinal Epithelial Cell Triggers Lipid Droplet Formation, Cytokine Production and Increased Cell Proliferation. Cell Cycle (2015) 14(16):2667–76. doi:10.1080/15384101.2015.1041684
104. Lee, S-M, Choi, H-J, Oh, C-H, Oh, J-W, and Han, J-S. Leptin Increases TNF-α Expression and Production through Phospholipase D1 in Raw 264.7 Cells. PloS one (2014) 9(7):e102373. doi:10.1371/journal.pone.0102373
105. Wu, D, Cheng, J, Sun, G, Wu, S, Li, M, Gao, Z, et al. p70S6K Promotes IL-6-induced Epithelial-Mesenchymal Transition and Metastasis of Head and Neck Squamous Cell Carcinoma. Oncotarget (2016) 7(24):36539–50. doi:10.18632/oncotarget.9282
106. Hao, Y, Baker, D, and Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Ijms (2019) 20(11):2767. doi:10.3390/ijms20112767
107. Xing, S, Yu, W, Zhang, X, Luo, Y, Lei, Z, Huang, D, et al. Isoviolanthin Extracted from Dendrobium Officinale Reverses TGF-Β1-Mediated Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma Cells via Deactivating the TGF-β/Smad and PI3K/Akt/mTOR Signaling Pathways. Ijms (2018) 19(6):1556. doi:10.3390/ijms19061556
108. Ogunwobi, OO, Wang, T, Zhang, L, and Liu, C. Cyclooxygenase-2 and Akt Mediate Multiple Growth-Factor-Induced Epithelial-Mesenchymal Transition in Human Hepatocellular Carcinoma. J Gastroenterol Hepatol (2012) 27(3):566–78. doi:10.1111/j.1440-1746.2011.06980.x
109. Bhat, AA, Ahmad, R, Uppada, SB, Singh, AB, and Dhawan, P. Claudin-1 Promotes TNF-α-Induced Epithelial-Mesenchymal Transition and Migration in Colorectal Adenocarcinoma Cells. Exp Cel Res (2016) 349(1):119–27. doi:10.1016/j.yexcr.2016.10.005
110. Wang, H, Wang, H-S, Zhou, B-H, Li, C-L, Zhang, F, Wang, X-F, et al. Epithelial-mesenchymal Transition (EMT) Induced by TNF-α Requires AKT/GSK-3β-mediated Stabilization of Snail in Colorectal Cancer. PloS one (2013) 8(2):e56664. doi:10.1371/journal.pone.0056664
111. Valle, A, Sastre-Serra, J, Pol, C, Miró, AM, Oliver, J, and Roca, P. Proteomic Analysis of MCF-7 Breast Cancer Cell Line Exposed to Leptin. Anal Cell Pathol (2011) 34(3):147–57. doi:10.1155/2011/405253
112. Katsogiannou, M, Andrieu, C, and Rocchi, P. Heat Shock Protein 27 Phosphorylation State Is Associated with Cancer Progression. Front Genet (2014) 5:346. doi:10.3389/fgene.2014.00346
113. Mendonsa, AM, Chalfant, MC, Gorden, LD, and VanSaun, MN. Modulation of the Leptin Receptor Mediates Tumor Growth and Migration of Pancreatic Cancer Cells. PloS one (2015) 10(4):e0126686. doi:10.1371/journal.pone.0126686
114. Giordano, C, Vizza, D, Panza, S, Barone, I, Bonofiglio, D, Lanzino, M, et al. Leptin Increases HER2 Protein Levels through a STAT3-Mediated Up-Regulation of Hsp90 in Breast Cancer Cells. Mol Oncol (2013) 7(3):379–91. doi:10.1016/j.molonc.2012.11.002
115. Lipsey, CC, Harbuzariu, A, Robey, RW, Huff, LM, Gottesman, MM, and Gonzalez-Perez, RR. Leptin Signaling Affects Survival and Chemoresistance of Estrogen Receptor Negative Breast Cancer. Ijms (2020) 21(11):3794. doi:10.3390/ijms21113794
116. Fiedor, E, Zajda, K, and Gregoraszczuk, EL. Leptin Receptor Antagonists' Action on HDAC Expression Eliminating the Negative Effects of Leptin in Ovarian Cancer. Cancer Genomics Proteomics (2018) 15(4):329–36. doi:10.21873/cgp.20091
117. Pachynski, RK, Wang, P, Salazar, N, Zheng, Y, Nease, L, Rosalez, J, et al. Chemerin Suppresses Breast Cancer Growth by Recruiting Immune Effector Cells into the Tumor Microenvironment. Front Immunol (2019) 10:983. doi:10.3389/fimmu.2019.00983
118. Harbuzariu, A, and Gonzalez-Perez, RR. Leptin-Notch axis Impairs 5-fluorouracil Effects on Pancreatic Cancer. Oncotarget (2018) 9(26):18239–53. doi:10.18632/oncotarget.24435
119. Osaki, M, and Okada, F. Exosomes and Their Role in Cancer Progression. Yonago Acta Med (2019) 62(2):182–90. doi:10.33160/yam.2019.06.002
120. Giordano, C, Gelsomino, L, Barone, I, Panza, S, Augimeri, G, Bonofiglio, D, et al. Leptin Modulates Exosome Biogenesis in Breast Cancer Cells: An Additional Mechanism in Cell-To-Cell Communication. Jcm (2019) 8(7):1027. doi:10.3390/jcm8071027
121. Raut, PK, Choi, DY, Kim, SH, Hong, JT, Kwon, TK, Jeong, JH, et al. Estrogen Receptor Signaling Mediates Leptin-Induced Growth of Breast Cancer Cells via Autophagy Induction. Oncotarget (2017) 8(65):109417–35. doi:10.18632/oncotarget.22684
122. Nepal, S, Kim, MJ, Hong, JT, Kim, SH, Sohn, D-H, Lee, SH, et al. Autophagy Induction by Leptin Contributes to Suppression of Apoptosis in Cancer Cells and Xenograft Model: Involvement of p53/FoxO3A axis. Oncotarget (2015) 6(9):7166–81. doi:10.18632/oncotarget.3347
123. Shrestha, M, and Park, P-H. p53 Signaling Is Involved in Leptin-Induced Growth of Hepatic and Breast Cancer Cells. Korean J Physiol Pharmacol (2016) 20(5):487–98. doi:10.4196/kjpp.2016.20.5.487
124. Yehuda-Shnaidman, E, Nimri, L, Tarnovscki, T, Kirshtein, B, Rudich, A, and Schwartz, B. Secreted Human Adipose Leptin Decreases Mitochondrial Respiration in HCT116 colon Cancer Cells. PloS one (2013) 8(9):e74843. doi:10.1371/journal.pone.0074843
125. Park, J, Kusminski, CM, Chua, SC, and Scherer, PE. Leptin Receptor Signaling Supports Cancer Cell Metabolism through Suppression of Mitochondrial Respiration In Vivo. Am J Pathol (2010) 177(6):3133–44. doi:10.2353/ajpath.2010.100595
126. Wang, C-Q, Wang, Y, Huang, B-F, Tang, C-H, Du, Z, Zeng, Y, et al. High Expression of Both Resistin and Fascin-1 Predicts a Poor Prognosis in Patients with Colorectal Cancer. Biomed Research International (2020) 2020:8753175. doi:10.1155/2020/8753175
127. Mihajlovic, M, Ninic, A, Sopic, M, Miljkovic, M, Stefanovic, A, Vekic, J, et al. Association Among Resistin, Adenylate Cyclase-Associated Protein 1 and High-Density Lipoprotein Cholesterol in Patients with Colorectal Cancer: a Multi-Marker Approach, as a Hallmark of Innovative Predictive, Preventive, and Personalized Medicine. EPMA J (2019) 10(3):307–16. doi:10.1007/s13167-019-00178-x
128. Avtanski, D, Garcia, A, Caraballo, B, Thangeswaran, P, Marin, S, Bianco, J, et al. Resistin Induces Breast Cancer Cells Epithelial to Mesenchymal Transition (EMT) and Stemness through Both Adenylyl Cyclase-Associated Protein 1 (CAP1)-dependent and CAP1-independent Mechanisms. Cytokine (2019) 120:155–64. doi:10.1016/j.cyto.2019.04.016
129. Weber, D, Satlof, L, Lavi, A, Bahl, K, Kaiser, M, Chen, K, et al. SAT-335 Resistin Induces Epithelial to Mesenchymal Transition (EMT) in Breast Cancer Cells through Activation of AXL Tyrosine Kinase Receptor. J Endocr Soc (2019) 3(Suppl. 1):SAT-335. doi:10.1210/js.2019-sat-335
130. Qiu, L, Zhang, G-F, Yu, L, Wang, H-Y, Jia, X-J, and Wang, T-J. Novel Oncogenic and Chemoresistance-Inducing Functions of Resistin in Ovarian Cancer Cells Require miRNAs-Mediated Induction of Epithelial-To-Mesenchymal Transition. Scientific Rep (2018) 8(1):12522. doi:10.1038/s41598-018-30978-6
131. Yang, C-C, Chang, S-F, Chao, J-K, Lai, Y-L, Chang, W-E, Hsu, W-H, et al. Activation of AMP-Activated Protein Kinase Attenuates Hepatocellular Carcinoma Cell Adhesion Stimulated by Adipokine Resistin. BMC cancer (2014) 14:112. doi:10.1186/1471-2407-14-112
132. Huang, W-S, Yang, J-T, Lu, C-C, Chang, S-F, Chen, C-N, Su, Y-P, et al. Fulvic Acid Attenuates Resistin-Induced Adhesion of HCT-116 Colorectal Cancer Cells to Endothelial Cells. Ijms (2015) 16(12):29370–82. doi:10.3390/ijms161226174
133. Hsieh, Y-Y, Shen, C-H, Huang, W-S, Chin, C-C, Kuo, Y-H, Hsieh, M, et al. Resistin-induced Stromal Cell-Derived Factor-1 Expression through Toll-like Receptor 4 and Activation of P38 MAPK/NFκB Signaling Pathway in Gastric Cancer Cells. J Biomed Sci (2014) 21(1):59. doi:10.1186/1423-0127-21-59
134. Deshmukh, SK, Srivastava, SK, Zubair, H, Bhardwaj, A, Tyagi, N, Al-Ghadhban, A, et al. Resistin Potentiates Chemoresistance and Stemness of Breast Cancer Cells: Implications for Racially Disparate Therapeutic Outcomes. Cancer Lett (2017) 396:21–9. doi:10.1016/j.canlet.2017.03.010
135. Mohammadi, M, Hedayati, M, Zarghami, N, and Ghaemmaghami, S. RESISTIN EFFECT ON TELOMERASE GENE EXPRESSION IN GASTRIC CANCER CELL LINE AGS. Acta Endo (Buc) (2016) 12(2):145–9. doi:10.4183/aeb.2016.145
136. Malvi, P, Chaube, B, Singh, SV, Mohammad, N, Vijayakumar, MV, Singh, S, et al. Elevated Circulatory Levels of Leptin and Resistin Impair Therapeutic Efficacy of Dacarbazine in Melanoma under Obese State. Cancer Metab (2018) 6(1):2. doi:10.1186/s40170-018-0176-5
137. Liu, Z, Shi, A, Song, D, Han, B, Zhang, Z, Ma, L, et al. Resistin Confers Resistance to Doxorubicin-Induced Apoptosis in Human Breast Cancer Cells through Autophagy Induction. Am J Cancer Res (2017) 7(3):574–83.
138. Kim, JG, Kim, EO, Jeong, BR, Min, YJ, Park, JW, Kim, ES, et al. Visfatin Stimulates Proliferation of MCF-7 Human Breast Cancer Cells. Mol Cell (2010) 30(4):341–5. doi:10.1007/s10059-010-0124-x
139. Buldak, RJ, Buldak, L, Polaniak, R, Kukla, M, Birkner, E, Kubina, R, et al. Visfatin Affects Redox Adaptative Responses and Proliferation in Me45 Human Malignant Melanoma Cells: an In Vitro Study. Oncol Rep (2013) 29(2):771–8.
140. Zhao, H, Tang, W, Chen, X, Wang, S, Wang, X, Xu, H, et al. The NAMPT/E2F2/SIRT1 axis Promotes Proliferation and Inhibits P53-dependent Apoptosis in Human Melanoma Cells. Biochem Biophysical Res Commun (2017) 493(1):77–84. doi:10.1016/j.bbrc.2017.09.071
141. Adya, R, Tan, BK, Punn, A, Chen, J, and Randeva, HS. Visfatin Induces Human Endothelial VEGF and MMP-2/9 Production via MAPK and PI3K/Akt Signalling Pathways: Novel Insights into Visfatin-Induced Angiogenesis. Cardiovasc Res (2008) 78(2):356–65. doi:10.1093/cvr/cvm111
142. Wang, P, Xu, T-Y, Guan, Y-F, Su, D-F, Fan, G-R, and Miao, C-Y. Perivascular Adipose Tissue-Derived Visfatin Is a Vascular Smooth Muscle Cell Growth Factor: Role of Nicotinamide Mononucleotide. Cardiovasc Res (2009) 81(2):370–80. doi:10.1093/cvr/cvn288
143. Kim, S-R, Bae, S-K, Choi, K-S, Park, S-Y, Jun, HO, Lee, J-Y, et al. Visfatin Promotes Angiogenesis by Activation of Extracellular Signal-Regulated Kinase 1/2. Biochem Biophysical Res Commun (2007) 357(1):150–6. doi:10.1016/j.bbrc.2007.03.105
144. Liu, P, Li, H, Cepeda, J, Xia, Y, Kempf, JA, Ye, H, et al. Regulation of Inflammatory Cytokine Expression in Pulmonary Epithelial Cells by Pre-B-cell colony-enhancing Factor via a Nonenzymatic and AP-1-dependent Mechanism. J Biol Chem (2009) 284(40):27344–51. doi:10.1074/jbc.m109.002519
145. Nowell, MA, Richards, PJ, Fielding, CA, Ognjanovic, S, Topley, N, Williams, AS, et al. Regulation of Pre-B Cell colony-enhancing Factor by STAT-3-dependent Interleukin-6trans-Signaling: Implications in the Pathogenesis of Rheumatoid Arthritis. Arthritis Rheum (2006) 54(7):2084–95. doi:10.1002/art.21942
146. Park, H-J, Kim, S-R, Kim, SS, Wee, H-J, Bae, M-K, Ryu, MH, et al. Visfatin Promotes Cell and Tumor Growth by Upregulating Notch1 in Breast Cancer. Oncotarget (2014) 5(13):5087–99. doi:10.18632/oncotarget.2086
147. Lu, G-W, Wang, Q-J, Xia, M-M, and Qian, J. Elevated Plasma Visfatin Levels Correlate with Poor Prognosis of Gastric Cancer Patients. Peptides (2014) 58:60–4. doi:10.1016/j.peptides.2014.05.016
148. Yang, J, Zhang, K, Song, H, Wu, M, Li, J, Yong, Z, et al. Visfatin Is Involved in Promotion of Colorectal Carcinoma Malignancy through an Inducing EMT Mechanism. Oncotarget (2016) 7(22):32306–17. doi:10.18632/oncotarget.8615
149. Liu, T, Miao, Z, Jiang, J, Yuan, S, Fang, W, Li, B, et al. Visfatin Mediates SCLC Cells Migration across Brain Endothelial Cells through Upregulation of CCL2. Int J Mol Sci (2015) 16(5):11439–51. doi:10.3390/ijms160511439
150. Lim, SY, Yuzhalin, AE, Gordon-Weeks, AN, and Muschel, RJ. Targeting the CCL2-CCR2 Signaling axis in Cancer Metastasis. Oncotarget (2016) 7(19):28697–710. doi:10.18632/oncotarget.7376
151. Wang, G-J, Shen, N-J, Cheng, L, Yehan Fang, F, Huang, H, and Li, K-H. Visfatin Triggers the In Vitro Migration of Osteosarcoma Cells via Activation of NF-κB/IL-6 Signals. Eur J Pharmacol (2016) 791:322–30. doi:10.1016/j.ejphar.2016.08.029
152. Cao, Z, Liang, N, Yang, H, and Li, S. Visfatin Mediates Doxorubicin Resistance in Human Non-small-cell Lung Cancer via Akt-Mediated Up-Regulation of ABCC1. Cel Prolif (2017) 50(5):e12366. doi:10.1111/cpr.12366
153. Kay-Pong, Y, Chi, LA-Y, and Samuel, CM. Interactions of Omentin and Lactotransferrin in the Progression of Metastatic Ovarian Cancer. FASEB J (2019) 33(1_Suppl. ment):704.5.
154. Li, D, Zhao, X, Xiao, Y, Mei, H, Pu, J, Xiang, X, et al. Intelectin 1 Suppresses Tumor Progression and Is Associated with Improved Survival in Gastric Cancer. Oncotarget (2015) 6(18):16168–82. doi:10.18632/oncotarget.3753
155. Yeung, CLA, Co, NN, Onstad, M, Yeung, T-L, Leung, CS, Schmandt, R, et al. Abstract 4887: Omentin: A Novel Adipokine in the Omental Microenvironment Associated with Ovarian Cancer Progression. Cancer Res (2014) 74(19 Suppl. ment):4887.
156. Kenny, HA, Chiang, C-Y, White, EA, Schryver, EM, Habis, M, Romero, IL, et al. Mesothelial Cells Promote Early Ovarian Cancer Metastasis through Fibronectin Secretion. J Clin Invest (2014) 124(10):4614–28. doi:10.1172/jci74778
157. Hiyoshi, M, Tsuno, NH, Otani, K, Kawai, K, Nishikawa, T, Shuno, Y, et al. Adiponectin Receptor 2 Is Negatively Associated with Lymph Node Metastasis of Colorectal Cancer. Oncol Lett (2012) 3(4):756–60. doi:10.3892/ol.2012.583
158. Taliaferro-Smith, L, Nagalingam, A, Zhong, D, Zhou, W, Saxena, NK, and Sharma, D. LKB1 Is Required for Adiponectin-Mediated Modulation of AMPK-S6k axis and Inhibition of Migration and Invasion of Breast Cancer Cells. Oncogene (2009) 28(29):2621–33. doi:10.1038/onc.2009.129
159. Corradetti, MN, Inoki, K, Bardeesy, N, DePinho, RA, and Guan, K-L. Regulation of the TSC Pathway by LKB1: Evidence of a Molecular Link between Tuberous Sclerosis Complex and Peutz-Jeghers Syndrome. Genes Develop (2004) 18(13):1533–8. doi:10.1101/gad.1199104
160. Gwinn, DM, Shackelford, DB, Egan, DF, Mihaylova, MM, Mery, A, Vasquez, DS, et al. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol Cel (2008) 30(2):214–26. doi:10.1016/j.molcel.2008.03.003
161. Amaral, CL, Freitas, LB, Tamura, RE, Tavares, MR, Pavan, ICB, Bajgelman, MC, et al. S6Ks Isoforms Contribute to Viability, Migration, Docetaxel Resistance and Tumor Formation of Prostate Cancer Cells. BMC cancer (2016) 16:602. doi:10.1186/s12885-016-2629-y
162. Zhang, S, Hu, B, Lv, X, Chen, S, Liu, W, and Shao, Z. The Prognostic Role of Ribosomal Protein S6 Kinase 1 Pathway in Patients with Solid Tumors: A Meta-Analysis. Front Oncol (2019) 9(390). doi:10.3389/fonc.2019.00390
163. Mauro, L, Pellegrino, M, Giordano, F, Ricchio, E, Rizza, P, De Amicis, F, et al. Estrogen Receptor‐α Drives Adiponectin Effects on Cyclin D1 Expression in Breast Cancer Cells. FASEB j. (2015) 29(5):2150–60. doi:10.1096/fj.14-262808
164. Wang, Y, Lam, JB, Lam, KSL, Liu, J, Lam, MC, Hoo, RLC, et al. Adiponectin Modulates the Glycogen Synthase Kinase-3β/β-Catenin Signaling Pathway and Attenuates Mammary Tumorigenesis of MDA-MB-231 Cells in Nude Mice. Cancer Res (2006) 66(23):11462–70. doi:10.1158/0008-5472.can-06-1969
165. Miyazaki, T, Bub, JD, Uzuki, M, and Iwamoto, Y. Adiponectin Activates C-Jun NH2-terminal Kinase and Inhibits Signal Transducer and Activator of Transcription 3. Biochem Biophysical Res Commun (2005) 333(1):79–87. doi:10.1016/j.bbrc.2005.05.076
166. Partida-Pérez, M, de la Luz Ayala-Madrigal, M, Peregrina-Sandoval, J, Macías-Gómez, N, Moreno-Ortiz, J, Leal-Ugarte, E, et al. Association of LEP and ADIPOQ Common Variants with Colorectal Cancer in Mexican Patients. Cancer Biomark (2010) 7(3):117–21. doi:10.3233/CBM-2010-0154
167. Li, G, Cong, L, Gasser, J, Zhao, J, Chen, K, and Li, F. Mechanisms Underlying the Anti-proliferative Actions of Adiponectin in Human Breast Cancer Cells, MCF7-Dependency on the cAMP/protein Kinase-A Pathway. Nutr Cancer (2011) 63(1):80–8. doi:10.1080/01635581.2010.516472
168. Körner, A, Pazaitou-Panayiotou, K, Kelesidis, T, Kelesidis, I, Williams, CJ, Kaprara, A, et al. Total and High-Molecular-Weight Adiponectin in Breast Cancer: In Vitro and In Vivo Studies. J Clin Endocrinol Metab (2007) 92(3):1041–8. doi:10.1210/jc.2006-1858
169. Cong, L, Gasser, J, Zhao, J, Yang, B, Li, F, and Zhao, AZ. Human Adiponectin Inhibits Cell Growth and Induces Apoptosis in Human Endometrial Carcinoma Cells, HEC-1-A and RL95-2. Endocr Relat Cancer (2007) 14(3):713–20. doi:10.1677/erc-07-0065
170. Dieudonne, M-N, Bussiere, M, Dos Santos, E, Leneveu, M-C, Giudicelli, Y, and Pecquery, R. Adiponectin Mediates Antiproliferative and Apoptotic Responses in Human MCF7 Breast Cancer Cells. Biochem Biophysical Res Commun (2006) 345(1):271–9. doi:10.1016/j.bbrc.2006.04.076
171. Zakikhani, M, Dowling, RJO, Sonenberg, N, and Pollak, MN. The Effects of Adiponectin and Metformin on Prostate and colon Neoplasia Involve Activation of AMP-Activated Protein Kinase. Cancer Prev Res (2008) 1(5):369–75. doi:10.1158/1940-6207.capr-08-0081
172. Fenton, JI, Birmingham, JM, Hursting, SD, and Hord, NG. Adiponectin Blocks Multiple Signaling Cascades Associated with Leptin-Induced Cell Proliferation inApcMin/+ colon Epithelial Cells. Int J Cancer (2008) 122(11):2437–45. doi:10.1002/ijc.23436
173. Jardé, T, Perrier, S, Vasson, M-P, and Caldefie-Chézet, F. Molecular Mechanisms of Leptin and Adiponectin in Breast Cancer. Eur J Cancer (2011) 47(1):33–43. doi:10.1016/j.ejca.2010.09.005
174. Luo, Z, Saha, AK, Xiang, X, and Ruderman, NB. AMPK, the Metabolic Syndrome and Cancer. Trends Pharmacol Sci (2005) 26(2):69–76. doi:10.1016/j.tips.2004.12.011
175. Kim, AY, Lee, YS, Kim, KH, Lee, JH, Lee, HK, Jang, S-H, et al. Adiponectin Represses Colon Cancer Cell Proliferation via AdipoR1- and -R2-Mediated AMPK Activation. Mol Endocrinol (2010) 24(7):1441–52. doi:10.1210/me.2009-0498
176. Kitajima, D, Kasamatsu, A, Nakashima, D, Miyamoto, I, Kimura, Y, Saito, T, et al. Tie2 Regulates Tumor Metastasis of Oral Squamous Cell Carcinomas. J Cancer (2016) 7(5):600–7. doi:10.7150/jca.13820
177. Kitajima, D, Kasamatsu, A, Nakashima, D, Miyamoto, I, Kimura, Y, Endo-Sakamoto, Y, et al. Evidence for Critical Role of Tie2/Ang1 Interaction in Metastatic Oral Cancer. Oncol Lett (2018) 15(5):7237–42. doi:10.3892/ol.2018.8212
178. Michael, IP, Orebrand, M, Lima, M, Pereira, B, Volpert, O, Quaggin, SE, et al. Angiopoietin-1 Deficiency Increases Tumor Metastasis in Mice. BMC cancer (2017) 17(1):539. doi:10.1186/s12885-017-3531-y
179. Wu, FT, Lee, CR, Bogdanovic, E, Prodeus, A, Gariépy, J, and Kerbel, RS. Vasculotide Reduces Endothelial Permeability and Tumor Cell Extravasation in the Absence of Binding to or Agonistic Activation of Tie2. EMBO Mol Med (2015) 7(6):770–87. doi:10.15252/emmm.201404193
180. Ou, X-L, Chen, H-J, Sun, W-H, Hang, C, Yang, L, Guan, Y-Y, et al. Effects of Angiopoietin-1 on Attachment and Metastasis Ofhuman Gastric Cancer Cell Line BGC-823. Wjg (2009) 15(43):5432–41. doi:10.3748/wjg.15.5432
181. Holopainen, T, Huang, H, Chen, C, Kim, KE, Zhang, L, Zhou, F, et al. Angiopoietin-1 Overexpression Modulates Vascular Endothelium to Facilitate Tumor Cell Dissemination and Metastasis Establishment. Cancer Res (2009) 69(11):4656–64. doi:10.1158/0008-5472.can-08-4654
182. Xu, Y, Liu, Y-j., and Yu, Q. Angiopoietin-3 Inhibits Pulmonary Metastasis by Inhibiting Tumor Angiogenesis. Cancer Res (2004) 64(17):6119–26. doi:10.1158/0008-5472.can-04-1054
183. Imanishi, Y, Hu, B, Jarzynka, MJ, Guo, P, Elishaev, E, Bar-Joseph, I, et al. Angiopoietin-2 Stimulates Breast Cancer Metastasis through the α5β1 Integrin-Mediated Pathway. Cancer Res (2007) 67(9):4254–63. doi:10.1158/0008-5472.can-06-4100
184. Li, C, Li, Q, Cai, Y, He, Y, Lan, X, Wang, W, et al. Overexpression of Angiopoietin 2 Promotes the Formation of Oral Squamous Cell Carcinoma by Increasing Epithelial-Mesenchymal Transition-Induced Angiogenesis. Cancer Gene Ther (2016) 23(9):295–302. doi:10.1038/cgt.2016.30
185. Han, HH, Kim, BG, Lee, JH, Kang, S, Kim, JE, and Cho, NH. Angiopoietin-2 Promotes ER+ Breast Cancer Cell Survival in Bone Marrow Niche. Endocr Relat Cancer (2016) 23(8):609–23. doi:10.1530/erc-16-0086
186. Sasaki, H, Suzuki, A, Shitara, M, Hikosaka, Y, Okuda, K, Moriyama, S, et al. Angiopoietin-like Protein ANGPTL2 Gene Expression Is Correlated with Lymph Node Metastasis in Lung Cancer. Oncol Lett (2012) 4(6):1325–8. doi:10.3892/ol.2012.924
187. Aoi, J, Endo, M, Kadomatsu, T, Miyata, K, Nakano, M, Horiguchi, H, et al. Angiopoietin-like Protein 2 Is an Important Facilitator of Inflammatory Carcinogenesis and Metastasis. Cancer Res (2011) 71(24):7502–12. doi:10.1158/0008-5472.can-11-1758
188. Endo, M, Nakano, M, Kadomatsu, T, Fukuhara, S, Kuroda, H, Mikami, S, et al. Tumor Cell-Derived Angiopoietin-like Protein ANGPTL2 Is a Critical Driver of Metastasis. Cancer Res (2012) 72(7):1784–94. doi:10.1158/0008-5472.can-11-3878
189. Wang, X, Hu, Z, Wang, Z, Cui, Y, and Cui, X. Angiopoietin-like Protein 2 Is an Important Facilitator of Tumor Proliferation, Metastasis, Angiogenesis and Glycolysis in Osteosarcoma. Am J Transl Res (2019) 11(10):6341–55.
190. Odagiri, H, Kadomatsu, T, Endo, M, Masuda, T, Morioka, MS, Fukuhara, S, et al. The Secreted Protein ANGPTL2 Promotes Metastasis of Osteosarcoma Cells through Integrin 5 1, P38 MAPK, and Matrix Metalloproteinases. Sci Signaling (2014) 7(309):ra7. doi:10.1126/scisignal.2004612
191. Galaup, A, Cazes, A, Le Jan, S, Philippe, J, Connault, E, Le Coz, E, et al. Angiopoietin-like 4 Prevents Metastasis through Inhibition of Vascular Permeability and Tumor Cell Motility and Invasiveness. Proc Natl Acad Sci (2006) 103(49):18721–6. doi:10.1073/pnas.0609025103
192. Zhang, H, Wong, CCL, Wei, H, Gilkes, DM, Korangath, P, Chaturvedi, P, et al. HIF-1-dependent Expression of Angiopoietin-like 4 and L1CAM Mediates Vascular Metastasis of Hypoxic Breast Cancer Cells to the Lungs. Oncogene (2012) 31(14):1757–70. doi:10.1038/onc.2011.365
193. Padua, D, Zhang, XH-F, Wang, Q, Nadal, C, Gerald, WL, Gomis, RR, et al. TGFβ Primes Breast Tumors for Lung Metastasis Seeding through Angiopoietin-like 4. Cell (2008) 133(1):66–77. doi:10.1016/j.cell.2008.01.046
194. Marchiò, S, Soster, M, Cardaci, S, Muratore, A, Bartolini, A, Barone, V, et al. A Complex of α 6 Integrin and E‐cadherin Drives Liver Metastasis of Colorectal Cancer Cells through Hepatic Angiopoietin‐like 6. EMBO Mol Med (2012) 4(11):1156–75. doi:10.1002/emmm.201101164
195. Chu, SH, Lee, MK, Ahn, KY, Im, JA, Park, MS, Lee, DC, et al. Chemerin and Adiponectin Contribute Reciprocally to Metabolic Syndrome. PLoS One (2012) 7(4):e34710. doi:10.1371/journal.pone.0034710
196. Li, J-J, Yin, H-K, Guan, D-X, Zhao, J-S, Feng, Y-X, Deng, Y-Z, et al. Chemerin Suppresses Hepatocellular Carcinoma Metastasis through CMKLR1-PTEN-Akt axis. Br J Cancer (2018) 118(10):1337–48. doi:10.1038/s41416-018-0077-y
197. Liu-Chittenden, Y, Jain, M, Gaskins, K, Wang, S, Merino, MJ, Kotian, S, et al. RARRES2 Functions as a Tumor Suppressor by Promoting β-catenin Phosphorylation/degradation and Inhibiting P38 Phosphorylation in Adrenocortical Carcinoma. Oncogene (2017) 36(25):3541–52. doi:10.1038/onc.2016.497
198. Zhang, J, Zhou, J, Tang, X, Zhou, L-Y, Zhai, L-L, Vanessa, ME-D, et al. Reduced Expression of Chemerin Is Associated with Poor Clinical Outcome in Acute Myeloid Leukemia. Oncotarget (2017) 8(54):92536–44. doi:10.18632/oncotarget.21440
199. Xu, C-H, Yang, Y, Wang, Y-C, Yan, J, and Qian, L-H. Prognostic Significance of Serum Chemerin Levels in Patients with Non-small Cell Lung Cancer. Oncotarget (2017) 8(14):22483–9. doi:10.18632/oncotarget.14956
200. Zhang, J, Jin, H-C, Zhu, A-K, Ying, R-C, Wei, W, and Zhang, F-J. Prognostic Significance of Plasma Chemerin Levels in Patients with Gastric Cancer. Peptides (2014) 61:7–11. doi:10.1016/j.peptides.2014.08.007
201. El-Sagheer, G, Gayyed, M, Ahmad, A, Abd El-Fattah, A, and Mohamed, M. Expression of Chemerin Correlates with a Poor Prognosis in Female Breast Cancer Patients. Breast Cancer (Dove Med Press) (2018) 10:169–76. doi:10.2147/bctt.s178181
202. De Henau, O, Degroot, G-N, Imbault, V, Robert, V, De Poorter, C, McHeik, S, et al. Signaling Properties of Chemerin Receptors CMKLR1, GPR1 and CCRL2. PloS one (2016) 11(10):e0164179. doi:10.1371/journal.pone.0164179
203. Duan, X, Kong, Z, Liu, Y, Zeng, Z, Li, S, Wu, W, et al. β-Arrestin2 Contributes to Cell Viability and Proliferation via the Down-Regulation of FOXO1 in Castration-Resistant Prostate Cancer. J Cel Physiol. (2015) 230(10):2371–81. doi:10.1002/jcp.24963
204. Ge, L, Shenoy, SK, Lefkowitz, RJ, and DeFea, K. Constitutive Protease-Activated Receptor-2-Mediated Migration of MDA MB-231 Breast Cancer Cells Requires Both β-Arrestin-1 and -2. J Biol Chem (2004) 279(53):55419–24. doi:10.1074/jbc.m410312200
205. Lakshmikanthan, V, Zou, L, Kim, JI, Michal, A, Nie, Z, Messias, NC, et al. Identification of Arrestin2 as a Corepressor of Androgen Receptor Signaling in Prostate Cancer. Proc Natl Acad Sci (2009) 106(23):9379–84. doi:10.1073/pnas.0900258106
206. Cong, L, Qiu, Z-y., Zhao, Y, Wang, W-b., Wang, C-x., Shen, H-c., et al. Loss of β-arrestin-2 and Activation of CXCR2 Correlate with Lymph Node Metastasis in Non-small Cell Lung Cancer. J Cancer (2017) 8(14):2785–92. doi:10.7150/jca.19631
207. Sun, W-Y, Hu, S-S, Wu, J-J, Huang, Q, Ma, Y, Wang, Q-T, et al. Down-regulation of β-arrestin2 Promotes Tumour Invasion and Indicates Poor Prognosis of Hepatocellular Carcinoma. Scientific Rep (2016) 6(1):35609. doi:10.1038/srep35609
208. DeFea, KA, Zalevsky, J, Thoma, MS, Déry, O, Mullins, RD, and Bunnett, NW. β-Arrestin-Dependent Endocytosis of Proteinase-Activated Receptor 2 Is Required for Intracellular Targeting of Activated Erk1/2. J Cel Biol (2000) 148(6):1267–82. doi:10.1083/jcb.148.6.1267
209. Rourke, JL, Dranse, HJ, and Sinal, CJ. CMKLR1 and GPR1 Mediate Chemerin Signaling through the RhoA/ROCK Pathway. Mol Cell Endocrinol (2015) 417:36–51. doi:10.1016/j.mce.2015.09.002
210. Yin, J, Lv, X, Hu, S, Zhao, X, Liu, Q, and Xie, H. Overexpression of Serum Response Factor Is Correlated with Poor Prognosis in Patients with Gastric Cancer. Hum Pathol (2019) 85:10–7. doi:10.1016/j.humpath.2018.10.018
211. O'Hurley, G, Prencipe, M, Lundon, D, O'Neill, A, Boyce, S, O'Grady, A, et al. The Analysis of Serum Response Factor Expression in Bone and Soft Tissue Prostate Cancer Metastases. Prostate (2014) 74(3):306–13. doi:10.1002/pros.22752
212. Cen, B, Selvaraj, A, Burgess, RC, Hitzler, JK, Ma, Z, Morris, SW, et al. Megakaryoblastic Leukemia 1, a Potent Transcriptional Coactivator for Serum Response Factor (SRF), Is Required for Serum Induction of SRF Target Genes. Mol Cel Biol (2003) 23(18):6597–608. doi:10.1128/mcb.23.18.6597-6608.2003
213. Selvaraj, A, and Prywes, R. Expression Profiling of Serum Inducible Genes Identifies a Subset of SRF Target Genes that Are MKL Dependent. BMC Mol Biol (2004) 5(1):13. doi:10.1186/1471-2199-5-13
214. Nair, P, Muthukkumar, S, Sells, SF, Han, S-S, Sukhatme, VP, and Rangnekar, VM. Early Growth Response-1-dependent Apoptosis Is Mediated by P53. J Biol Chem (1997) 272(32):20131–8. doi:10.1074/jbc.272.32.20131
215. Krones-Herzig, A, Adamson, E, and Mercola, D. Early Growth Response 1 Protein, an Upstream Gatekeeper of the P53 Tumor Suppressor, Controls Replicative Senescence. Proc Natl Acad Sci (2003) 100(6):3233–8. doi:10.1073/pnas.2628034100
216. Li, L, Huang, C, Zhang, X, Wang, J, Ma, P, Liu, Y, et al. Chemerin-derived Peptide C-20 Suppressed Gonadal Steroidogenesis. Am J Reprod Immunol (2014) 71(3):265–77. doi:10.1111/aji.12164
217. Muruganandan, S, Dranse, HJ, Rourke, JL, McMullen, NM, and Sinal, CJ. Chemerin Neutralization Blocks Hematopoietic Stem Cell Osteoclastogenesis. Stem Cells (2013) 31(10):2172–82. doi:10.1002/stem.1450
218. Guo, JC, Li, J, Zhao, YP, Zhou, L, Cui, QC, Zhou, WX, et al. Expression of C-Fos Was Associated with Clinicopathologic Characteristics and Prognosis in Pancreatic Cancer. PLoS One (2015) 10(3):e0120332. doi:10.1371/journal.pone.0120332
219. Lu, C, Shen, Q, DuPré, E, Kim, H, Hilsenbeck, S, and Brown, PH. cFos Is Critical for MCF-7 Breast Cancer Cell Growth. Oncogene (2005) 24(43):6516–24. doi:10.1038/sj.onc.1208905
220. Mikula, M, Gotzmann, J, Fischer, ANM, Wolschek, MF, Thallinger, C, Schulte-Hermann, R, et al. The Proto-Oncoprotein C-Fos Negatively Regulates Hepatocellular Tumorigenesis. Oncogene (2003) 22(43):6725–38. doi:10.1038/sj.onc.1206781
221. Mahner, S, Baasch, C, Schwarz, J, Hein, S, Wölber, L, Jänicke, F, et al. C-fos Expression Is a Molecular Predictor of Progression and Survival in Epithelial Ovarian Carcinoma. Br J Cancer (2008) 99(8):1269–75. doi:10.1038/sj.bjc.6604650
222. Oliveira-Ferrer, L, Rößler, K, Haustein, V, Schröder, C, Wicklein, D, Maltseva, D, et al. c-FOS Suppresses Ovarian Cancer Progression by Changing Adhesion. Br J Cancer (2014) 110(3):753–63. doi:10.1038/bjc.2013.774
223. Zhang, X, Zhang, L, Yang, H, Huang, X, Otu, H, Libermann, TA, et al. c-Fos as a Proapoptotic Agent in TRAIL-Induced Apoptosis in Prostate Cancer Cells. Cancer Res (2007) 67(19):9425–34. doi:10.1158/0008-5472.can-07-1310
224. Li, T, Guo, H, Song, Y, Zhao, X, Shi, Y, Lu, Y, et al. Loss of Vinculin and Membrane-Bound β-catenin Promotes Metastasis and Predicts Poor Prognosis in Colorectal Cancer. Mol Cancer (2014) 13(1):263. doi:10.1186/1476-4598-13-263
225. Zhang, M, Liu, P, Xu, F, He, Y, Xie, X, and Jiang, X. Retracted : Vinculin Promotes Gastric Cancer Proliferation and Migration and Predicts Poor Prognosis in Patients with Gastric Cancer. J Cel Biochem (2019) 120(8):14107–15. doi:10.1002/jcb.28686
226. Pan, Y, Zhou, F, He, C, Hui, L, Huang, T, and Wei, Y. Leptin-LepRb Expressed in Gastric Cancer Patients and Related to Cancer-Related Depression. Biomed Res Int (2017) 2017:6482842. doi:10.1155/2017/6482842
227. Cheng, H, Wang, Z, Fu, L, and Xu, T. Macrophage Polarization in the Development and Progression of Ovarian Cancers: An Overview. Front Oncol (2019) 9:421. doi:10.3389/fonc.2019.00421
228. Yafei, Z, Jun, G, and Guolan, G. Correlation between Macrophage Infiltration and Prognosis of Ovarian Cancer-A Preliminary Study. Biomed Res (2016) 27:305–12.
229. Tian, Z, Hou, X, Liu, W, Han, Z, and Wei, L. Macrophages and Hepatocellular Carcinoma. Cel Biosci (2019) 9(1):79. doi:10.1186/s13578-019-0342-7
230. Jeong, H, Hwang, I, Kang, SH, Shin, HC, and Kwon, SY. Tumor-Associated Macrophages as Potential Prognostic Biomarkers of Invasive Breast Cancer. J Breast Cancer (2019) 22(1):38–51. doi:10.4048/jbc.2019.22.e5
231. Lee, YS, Song, SJ, Hong, HK, Oh, BY, Lee, WY, and Cho, YB. The FBW7-MCL-1 axis Is Key in M1 and M2 Macrophage-Related colon Cancer Cell Progression: Validating the Immunotherapeutic Value of Targeting PI3Kγ. Exp Mol Med (2020) 52(5):815–31. doi:10.1038/s12276-020-0436-7
232. Herová, M, Schmid, M, Gemperle, C, and Hersberger, M. ChemR23, the Receptor for Chemerin and Resolvin E1, Is Expressed and Functional on M1 but Not on M2 Macrophages. J.I. (2015) 194(5):2330–7. doi:10.4049/jimmunol.1402166
233. Pachynski, RK, Zabel, BA, Kohrt, HE, Tejeda, NM, Monnier, J, Swanson, CD, et al. The Chemoattractant Chemerin Suppresses Melanoma by Recruiting Natural Killer Cell Antitumor Defenses. J Exp Med (2012) 209(8):1427–35. doi:10.1084/jem.20112124
234. Dimitriadis, GK, Kaur, J, Adya, R, Miras, AD, Mattu, HS, Hattersley, JG, et al. Chemerin Induces Endothelial Cell Inflammation: Activation of Nuclear Factor-Kappa Beta and Monocyte-Endothelial Adhesion. Oncotarget (2018) 9(24):16678–90. doi:10.18632/oncotarget.24659
235. Zylla, S, Pietzner, M, Kühn, J-P, Völzke, H, Dörr, M, Nauck, M, et al. Serum Chemerin Is Associated with Inflammatory and Metabolic Parameters-Results of a Population-Based Study. Obesity (2017) 25(2):468–75. doi:10.1002/oby.21735
236. Liu, S, Hempe, JM, McCarter, RJ, Li, S, and Fonseca, VA. Association between Inflammation and Biological Variation in Hemoglobin A1c in U.S. Nondiabetic Adults. J Clin Endocrinol Metab (2015) 100(6):2364–71. doi:10.1210/jc.2014-4454
237. Kaur, J, Adya, R, Tan, BK, Chen, J, and Randeva, HS. Identification of Chemerin Receptor (ChemR23) in Human Endothelial Cells: Chemerin-Induced Endothelial Angiogenesis. Biochem Biophysical Res Commun (2010) 391(4):1762–8. doi:10.1016/j.bbrc.2009.12.150
238. Bozaoglu, K, Curran, JE, Stocker, CJ, Zaibi, MS, Segal, D, Konstantopoulos, N, et al. Chemerin, a Novel Adipokine in the Regulation of Angiogenesis. J Clin Endocrinol Metab (2010) 95(5):2476–85. doi:10.1210/jc.2010-0042
239. Wang, C, Wu, WKK, Liu, X, To, K-F, Chen, GG, Yu, J, et al. Increased Serum Chemerin Level Promotes Cellular Invasiveness in Gastric Cancer: a Clinical and Experimental Study. Peptides (2014) 51:131–8. doi:10.1016/j.peptides.2013.10.009
240. Nakamura, N, Naruse, K, Kobayashi, Y, Miyabe, M, Saiki, T, Enomoto, A, et al. Chemerin Promotes Angiogenesis In Vivo. Physiol Rep (2018) 6(24):e13962. doi:10.14814/phy2.13962
241. Skrzeczynska-Moncznik, J, Wawro, K, Stefanska, A, Oleszycka, E, Kulig, P, Zabel, BA, et al. Potential Role of Chemerin in Recruitment of Plasmacytoid Dendritic Cells to Diseased Skin. Biochem biophysical Res Commun (2009) 380:323–7.
242. Bidwell, BN, Slaney, CY, Withana, NP, Forster, S, Cao, Y, Loi, S, et al. Silencing of Irf7 Pathways in Breast Cancer Cells Promotes Bone Metastasis through Immune Escape. Nat Med (2012) 18(8):1224–31. doi:10.1038/nm.2830
243. Bekeredjian-Ding, I, Schäfer, M, Hartmann, E, Pries, R, Parcina, M, Schneider, P, et al. Tumour-derived Prostaglandin E2and Transforming Growth Factor-β Synergize to Inhibit Plasmacytoid Dendritic Cell-Derived Interferon-α. Immunology (2009) 128(3):439–50. doi:10.1111/j.1365-2567.2009.03134.x
244. Hartmann, E, Wollenberg, B, Rothenfusser, S, Wagner, M, Wellisch, D, Mack, B, et al. Identification and Functional Analysis of Tumor-Infiltrating Plasmacytoid Dendritic Cells in Head and Neck Cancer. Cancer Res (2003) 63(19):6478–87.
245. Auguet, T, Quintero, Y, Terra, X, Martínez, S, Lucas, A, Pellitero, S, et al. Upregulation of Lipocalin 2 in Adipose Tissues of Severely Obese Women: Positive Relationship with Proinflammatory Cytokines. Obesity (Silver Spring) (2011) 19(12):2295–300. doi:10.1038/oby.2011.61
246. Lee, H-J, Lee, E-K, Lee, K-J, Hong, S-W, Yoon, Y, and Kim, J-S. Ectopic Expression of Neutrophil Gelatinase-Associated Lipocalin Suppresses the Invasion and Liver Metastasis of colon Cancer Cells. Int J Cancer (2006) 118(10):2490–7. doi:10.1002/ijc.21657
247. Feng, M, Feng, J, Chen, W, Wang, W, Wu, X, Zhang, J, et al. Lipocalin2 Suppresses Metastasis of Colorectal Cancer by Attenuating NF-κb-dependent Activation of Snail and Epithelial Mesenchymal Transition. Mol Cancer (2016) 15(1):77. doi:10.1186/s12943-016-0564-9
248. Moniaux, N, Chakraborty, S, Yalniz, M, Gonzalez, J, Shostrom, VK, Standop, J, et al. Early Diagnosis of Pancreatic Cancer: Neutrophil Gelatinase-Associated Lipocalin as a Marker of Pancreatic Intraepithelial Neoplasia. Br J Cancer (2008) 98(9):1540–7. doi:10.1038/sj.bjc.6604329
249. Yang, J, Bielenberg, DR, Rodig, SJ, Doiron, R, Clifton, MC, Kung, AL, et al. Lipocalin 2 Promotes Breast Cancer Progression. Proc Natl Acad Sci (2009) 106(10):3913–8. doi:10.1073/pnas.0810617106
250. Zhang, Y, Lazarus, J, Steele, NG, Yan, W, Lee, H-J, Nwosu, ZC, et al. Regulatory T-Cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov (2020) 10(3):422–39. doi:10.1158/2159-8290.cd-19-0958
251. Karlsen, JR, Borregaard, N, and Cowland, JB. Induction of Neutrophil Gelatinase-Associated Lipocalin Expression by Co-stimulation with Interleukin-17 and Tumor Necrosis Factor-α Is Controlled by IκB-ζ but Neither by C/EBP-β Nor C/EBP-δ. J Biol Chem (2010) 285(19):14088–100. doi:10.1074/jbc.m109.017129
252. Wani, NA, Nasser, MW, Ahirwar, DK, Zhao, H, Miao, Z, Shilo, K, et al. C-X-C Motif Chemokine 12/C-X-C Chemokine Receptor Type 7 Signaling Regulates Breast Cancer Growth and Metastasis by Modulating the Tumor Microenvironment. Breast Cancer Res (2014) 16(3):R54. doi:10.1186/bcr3665
253. Gomez-Chou, SB, Swidnicka-Siergiejko, AK, Badi, N, Chavez-Tomar, M, Lesinski, GB, Bekaii-Saab, T, et al. Lipocalin-2 Promotes Pancreatic Ductal Adenocarcinoma by Regulating Inflammation in the Tumor Microenvironment. Cancer Res (2017) 77(10):2647–60. doi:10.1158/0008-5472.can-16-1986
254. Tschesche, H, Zölzer, V, Triebel, S, and Bartsch, S. The Human Neutrophil Lipocalin Supports the Allosteric Activation of Matrix Metalloproteinases. Eur J Biochem (2001) 268(7):1918–28. doi:10.1046/j.1432-1327.2001.02066.x
255. Kubben, FJGM, Sier, CFM, Hawinkels, LJAC, Tschesche, H, van Duijn, W, Zuidwijk, K, et al. Clinical Evidence for a Protective Role of Lipocalin-2 against MMP-9 Autodegradation and the Impact for Gastric Cancer. Eur J Cancer (2007) 43(12):1869–76. doi:10.1016/j.ejca.2007.05.013
256. Nuntagowat, C, Leelawat, K, and Tohtong, R. NGAL Knockdown by siRNA in Human Cholangiocarcinoma Cells Suppressed Invasion by Reducing NGAL/MMP-9 Complex Formation. Clin Exp Metastasis (2010) 27(5):295–305. doi:10.1007/s10585-010-9327-y
257. Fernández, CA, Yan, L, Louis, G, Yang, J, Kutok, JL, and Moses, MA. The Matrix Metalloproteinase-9/neutrophil Gelatinase-Associated Lipocalin Complex Plays a Role in Breast Tumor Growth and Is Present in the Urine of Breast Cancer Patients. Clin Cancer Res (2005) 11(15):5390–5. doi:10.1158/1078-0432.ccr-04-2391
258. Volpe, V, Raia, Z, Sanguigno, L, Somma, D, Mastrovito, P, Moscato, F, et al. NGAL Controls the Metastatic Potential of Anaplastic Thyroid Carcinoma Cells. J Clin Endocrinol Metab (2013) 98(1):228–35. doi:10.1210/jc.2012-2528
259. Roy, R, Louis, G, Loughlin, KR, Wiederschain, D, Kilroy, SM, Lamb, CC, et al. Tumor-specific Urinary Matrix Metalloproteinase Fingerprinting: Identification of High Molecular Weight Urinary Matrix Metalloproteinase Species. Clin Cancer Res (2008) 14(20):6610–7. doi:10.1158/1078-0432.ccr-08-1136
260. Leng, X, Ding, T, Lin, H, Wang, Y, Hu, L, Hu, J, et al. Inhibition of Lipocalin 2 Impairs Breast Tumorigenesis and Metastasis. Cancer Res (2009) 69(22):8579–84. doi:10.1158/0008-5472.can-09-1934
261. Iannetti, A, Pacifico, F, Acquaviva, R, Lavorgna, A, Crescenzi, E, Vascotto, C, et al. The Neutrophil Gelatinase-Associated Lipocalin (NGAL), a NF- B-Regulated Gene, Is a Survival Factor for Thyroid Neoplastic Cells. Proc Natl Acad Sci (2008) 105(37):14058–63. doi:10.1073/pnas.0710846105
262. Leung, L, Radulovich, N, Zhu, CQ, Organ, S, Bandarchi, B, Pintilie, M, et al. Lipocalin2 Promotes Invasion, Tumorigenicity and Gemcitabine Resistance in Pancreatic Ductal Adenocarcinoma. PLoS One (2012) 7(10):e46677. doi:10.1371/journal.pone.0046677
263. Hanai, J-i., Mammoto, T, Seth, P, Mori, K, Karumanchi, SA, Barasch, J, et al. Lipocalin 2 Diminishes Invasiveness and Metastasis of Ras-Transformed Cells. J Biol Chem (2005) 280(14):13641–7. doi:10.1074/jbc.m413047200
264. Shi, H, Gu, Y, Yang, J, Xu, L, Mi, W, and Yu, W. Lipocalin 2 Promotes Lung Metastasis of Murine Breast Cancer Cells. J Exp Clin Cancer Res (2008) 27(1):83. doi:10.1186/1756-9966-27-83
265. Gu, Y, Zhang, J, Mi, W, Yang, J, Han, F, Lu, X, et al. Silencing of GM3 Synthase Suppresses Lung Metastasis of Murine Breast Cancer Cells. Breast Cancer Res (2008) 10(1):R1. doi:10.1186/bcr1841
266. Li, B, Xu, WW, Lam, AKY, Wang, Y, Hu, H-F, Guan, XY, et al. Significance of PI3K/AKT Signaling Pathway in Metastasis of Esophageal Squamous Cell Carcinoma and its Potential as a Target for Anti-metastasis Therapy. Oncotarget (2017) 8(24):38755–66. doi:10.18632/oncotarget.16333
267. Yue, Y, Hui, K, Wu, S, Zhang, M, Que, T, Gu, Y, et al. MUC15 Inhibits Cancer Metastasis via PI3K/AKT Signaling in Renal Cell Carcinoma. Cel Death Dis (2020) 11(5):336. doi:10.1038/s41419-020-2518-9
268. Liu, H, Radisky, DC, Nelson, CM, Zhang, H, Fata, JE, Roth, RA, et al. Mechanism of Akt1 Inhibition of Breast Cancer Cell Invasion Reveals a Protumorigenic Role for TSC2. Proc Natl Acad Sci (2006) 103(11):4134–9. doi:10.1073/pnas.0511342103
269. Qin, J-J, Li, X, Wang, W, Zi, X, and Zhang, R. Targeting the NFAT1-MDM2-MDMX Network Inhibits the Proliferation and Invasion of Prostate Cancer Cells, Independent of P53 and Androgen. Front Pharmacol (2017) 8:917. doi:10.3389/fphar.2017.00917
270. Bao, G, Clifton, M, Hoette, TM, Mori, K, Deng, S-X, Qiu, A, et al. Iron Traffics in Circulation Bound to a Siderocalin (Ngal)-Catechol Complex. Nat Chem Biol (2010) 6(8):602–9. doi:10.1038/nchembio.402
271. Schafer, ZT, Grassian, AR, Song, L, Jiang, Z, Gerhart-Hines, Z, Irie, HY, et al. Antioxidant and Oncogene rescue of Metabolic Defects Caused by Loss of Matrix Attachment. Nature (2009) 461(7260):109–13. doi:10.1038/nature08268
272. Devireddy, LR, Gazin, C, Zhu, X, and Green, MR. A Cell-Surface Receptor for Lipocalin 24p3 Selectively Mediates Apoptosis and Iron Uptake. Cell (2005) 123(7):1293–305. doi:10.1016/j.cell.2005.10.027
273. Zhang, S, Chen, Y, Guo, W, Yuan, L, Zhang, D, Xu, Y, et al. Disordered Hepcidin-Ferroportin Signaling Promotes Breast Cancer Growth. Cell Signal (2014) 26(11):2539–50. doi:10.1016/j.cellsig.2014.07.029
274. Deng, Y, Wang, ZV, Gordillo, R, An, Y, Zhang, C, Liang, Q, et al. An Adipo-Biliary-Uridine axis that Regulates Energy Homeostasis. Science (2017) 355(6330). doi:10.1126/science.aaf5375
275. Kohli, R, Bhattacharjee, J, and Inge, TH. Postprandial Uridine Physiology Is Altered by Obesity. Gastroenterology (2018) 155(5):1645–6. doi:10.1053/j.gastro.2018.07.043
276. Placet, M, Arguin, G, Molle, CM, Babeu, J-P, Jones, C, Carrier, JC, et al. The G Protein-Coupled P2Y6 Receptor Promotes Colorectal Cancer Tumorigenesis by Inhibiting Apoptosis. Biochim Biophys Acta (Bba) - Mol Basis Dis (2018) 1864(5):1539–51. doi:10.1016/j.bbadis.2018.02.008
277. Qiu, Y, Liu, Y, Li, WH, Zhang, HQ, Tian, XX, and Fang, WG. P2Y2 Receptor Promotes the Migration and Invasion of Breast Cancer Cells via EMT-Related Genes Snail and E-Cadherin. Oncol Rep (2018) 39(1):138–50. doi:10.3892/or.2017.6081
278. Ma, X, Pan, X, Wei, Y, Tan, B, Yang, L, Ren, H, et al. Chemotherapy-induced Uridine Diphosphate Release Promotes Breast Cancer Metastasis through P2Y6 Activation. Oncotarget (2016) 7(20):29036–50. doi:10.18632/oncotarget.8664
279. Miyashita, H, Takebayashi, Y, Eliason, JF, Fujimori, F, Nitta, Y, Sato, A, et al. Uridine Phosphorylase Is a Potential Prognostic Factor in Patients with Oral Squamous Cell Carcinoma. Cancer (2002) 94(11):2959–66. doi:10.1002/cncr.10568
280. Shen, G, He, P, Mao, Y, Li, P, Luh, F, Ding, G, et al. Overexpression of Uridine-Cytidine Kinase 2 Correlates with Breast Cancer Progression and Poor Prognosis. J Breast Cancer (2017) 20(2):132–41. doi:10.4048/jbc.2017.20.2.132
281. Guan, Y, Bhandari, A, Zhang, X, and Wang, O. Uridine Phosphorylase 1 Associates to Biological and Clinical Significance in Thyroid Carcinoma Cell Lines. J Cel Mol Med (2019) 23(11):7438–48. doi:10.1111/jcmm.14612
282. Wu, Y, Jamal, M, Xie, T, Sun, J, Song, T, Yin, Q, et al. Uridine‐cytidine Kinase 2 (UCK2): A Potential Diagnostic and Prognostic Biomarker for Lung Cancer. Cancer Sci (2019) 110(9):2734–47. doi:10.1111/cas.14125
283. Mazidi, M, Katsiki, N, Mikhailidis, DP, and Banach, M. The Link between Insulin Resistance Parameters and Serum Uric Acid Is Mediated by Adiposity. Atherosclerosis (2018) 270:180–6. doi:10.1016/j.atherosclerosis.2017.12.033
284. Mele, C, Tagliaferri, MA, Saraceno, G, Mai, S, Vietti, R, Zavattaro, M, et al. Serum Uric Acid Potentially Links Metabolic Health to Measures of Fuel Use in Lean and Obese Individuals. Nutr Metab Cardiovasc Dis (2018) 28(10):1029–35. doi:10.1016/j.numecd.2018.06.010
285. Ali, N, Perveen, R, Rahman, S, Mahmood, S, Rahman, S, Islam, S, et al. Prevalence of Hyperuricemia and the Relationship between Serum Uric Acid and Obesity: A Study on Bangladeshi Adults. PloS one (2018) 13(11):e0206850. doi:10.1371/journal.pone.0206850
286. Levine, W, Dyer, AR, Shekelle, RB, Schoenberger, JA, and Stamler, J. Serum Uric Acid and 11.5-year Mortality of Middle-Aged Women: Findings of the Chicago Heart Association Detection Project in Industry. J Clin Epidemiol (1989) 42(3):257–67. doi:10.1016/0895-4356(89)90061-9
287. Kolonel, LN, Yoshizawa, C, Nomura, AM, and Stemmermann, GN. Relationship of Serum Uric Acid to Cancer Occurrence in a Prospective Male Cohort. Cancer Epidemiol Biomarkers Prev (1994) 3:225–8.
288. Yan, S, Zhang, P, Xu, W, Liu, Y, Wang, B, Jiang, T, et al. Serum Uric Acid Increases Risk of Cancer Incidence and Mortality: A Systematic Review and Meta-Analysis. Mediators Inflamm (2015) 2015:764250. doi:10.1155/2015/764250
289. Hammarsten, J, Damber, J-E, Peeker, R, Mellström, D, and Högstedt, B. A Higher Prediagnostic Insulin Level Is a Prospective Risk Factor for Incident Prostate Cancer. Cancer Epidemiol (2010) 34(5):574–9. doi:10.1016/j.canep.2010.06.014
290. Yim, K, Bindayi, A, McKay, R, Mehrazin, R, Raheem, OA, Field, C, et al. Rising Serum Uric Acid Level Is Negatively Associated with Survival in Renal Cell Carcinoma. Cancers (2019) 11(4):536. doi:10.3390/cancers11040536
291. Cetin, AO, Omar, M, Calp, S, Tunca, H, Yimaz, N, Ozseker, B, et al. Hyperuricemia at the Time of Diagnosis Is a Factor for Poor Prognosis in Patients with Stage II and III Colorectal Cancer (Uric Acid and Colorectal Cancer). Asian Pac J Cancer Prev (2017) 18(2):485–90. doi:10.22034/APJCP.2017.18.2.485
292. Subandrate, S, Lea, K, and Safyudin, . The Correlation between Uric Acid and Stages of Malignancy Among Gastric Cancer Patient in Palembang, Indonesia. J Phys Conf Ser (2019) 1246:012062. doi:10.1088/1742-6596/1246/1/012062
293. Wang, S, Liu, X, He, Z, Chen, X, and Li, W. Hyperuricemia Has an Adverse Impact on the Prognosis of Patients with Osteosarcoma. Tumor Biol (2016) 37(1):1205–10. doi:10.1007/s13277-015-3830-3
294. Fabbrini, E, Serafini, M, Colic Baric, I, Hazen, SL, and Klein, S. Effect of Plasma Uric Acid on Antioxidant Capacity, Oxidative Stress, and Insulin Sensitivity in Obese Subjects. Diabetes (2014) 63(3):976–81. doi:10.2337/db13-1396
295. Gersch, C, Palii, SP, Kim, KM, Angerhofer, A, Johnson, RJ, and Henderson, GN. Inactivation of Nitric Oxide by Uric Acid. Nucleosides, Nucleotides and Nucleic Acids (2008) 27(8):967–78. doi:10.1080/15257770802257952
296. Wan, X, Xu, C, Lin, Y, Lu, C, Li, D, Sang, J, et al. Uric Acid Regulates Hepatic Steatosis and Insulin Resistance through the NLRP3 Inflammasome-dependent Mechanism. J Hepatol (2016) 64(4):925–32. doi:10.1016/j.jhep.2015.11.022
297. Sautin, YY, Nakagawa, T, Zharikov, S, and Johnson, RJ. Adverse Effects of the Classic Antioxidant Uric Acid in Adipocytes: NADPH Oxidase-Mediated Oxidative/nitrosative Stress. Am J Physiology-Cell Physiol (2007) 293(2):C584–C596. doi:10.1152/ajpcell.00600.2006
298. Yu, M-A, Sánchez-Lozada, LG, Johnson, RJ, and Kang, D-H. Oxidative Stress with an Activation of the Renin-Angiotensin System in Human Vascular Endothelial Cells as a Novel Mechanism of Uric Acid-Induced Endothelial Dysfunction. J Hypertens (2010) 28(6):1234–42. doi:10.1097/hjh.0b013e328337da1d
299. Corry, DB, Eslami, P, Yamamoto, K, Nyby, MD, Makino, H, and Tuck, ML. Uric Acid Stimulates Vascular Smooth Muscle Cell Proliferation and Oxidative Stress via the Vascular Renin-Angiotensin System. J Hypertens (2008) 26(2):269–75. doi:10.1097/hjh.0b013e3282f240bf
300. Cirillo, P, Gersch, MS, Mu, W, Scherer, PM, Kim, KM, Gesualdo, L, et al. Ketohexokinase-dependent Metabolism of Fructose Induces Proinflammatory Mediators in Proximal Tubular Cells. Jasn (2009) 20(3):545–53. doi:10.1681/asn.2008060576
301. Lanaspa, MA, Sanchez-Lozada, LG, Choi, Y-J, Cicerchi, C, Kanbay, M, Roncal-Jimenez, CA, et al. Uric Acid Induces Hepatic Steatosis by Generation of Mitochondrial Oxidative Stress. J Biol Chem (2012) 287(48):40732–44. doi:10.1074/jbc.m112.399899
302. DeNicola, GM, Karreth, FA, Humpton, TJ, Gopinathan, A, Wei, C, Frese, K, et al. Oncogene-induced Nrf2 Transcription Promotes ROS Detoxification and Tumorigenesis. Nature (2011) 475(7354):106–9. doi:10.1038/nature10189
303. Ames, BN, Cathcart, R, Schwiers, E, and Hochstein, P. Uric Acid Provides an Antioxidant Defense in Humans against Oxidant- and Radical-Caused Aging and Cancer: a Hypothesis. Proc Natl Acad Sci (1981) 78(11):6858–62. doi:10.1073/pnas.78.11.6858
304. Linder, N, Haglund, C, Lundin, M, Nordling, S, Ristimäki, A, Kokkola, A, et al. Decreased Xanthine Oxidoreductase Is a Predictor of Poor Prognosis in Early-Stage Gastric Cancer. J Clin Pathol (2006) 59(9):965–71. doi:10.1136/jcp.2005.032524
305. Linder, N, Martelin, E, Lundin, M, Louhimo, J, Nordling, S, Haglund, C, et al. Xanthine Oxidoreductase - Clinical Significance in Colorectal Cancer and In Vitro Expression of the Protein in Human colon Cancer Cells. Eur J Cancer (2009) 45(4):648–55. doi:10.1016/j.ejca.2008.10.036
306. Linder, N, Lundin, J, Isola, J, Lundin, M, Raivio, KO, and Joensuu, H. Down-Regulated Xanthine Oxidoreductase Is a Feature of Aggressive Breast Cancer. Clin Cancer Res (2005) 11:4372–81. doi:10.1158/1078-0432.ccr-04-2280
307. Cheung, KJ, Tzameli, I, Pissios, P, Rovira, I, Gavrilova, O, Ohtsubo, T, et al. Xanthine Oxidoreductase Is a Regulator of Adipogenesis and PPARγ Activity. Cel Metab (2007) 5(2):115–28. doi:10.1016/j.cmet.2007.01.005
308. Fini, MA, Monks, J, Farabaugh, SM, and Wright, RM. Contribution of Xanthine Oxidoreductase to Mammary Epithelial and Breast Cancer Cell Differentiation in Part Modulates Inhibitor of Differentiation-1. Mol Cancer Res (2011) 9(9):1242–54. doi:10.1158/1541-7786.mcr-11-0176
309. Fini, MA, Orchard-Webb, D, Kosmider, B, Amon, JD, Kelland, R, Shibao, G, et al. Migratory Activity of Human Breast Cancer Cells Is Modulated by Differential Expression of Xanthine Oxidoreductase. J Cel Biochem (2008) 105(4):1008–26. doi:10.1002/jcb.21901
310. Chen, D, Forootan, SS, Gosney, JR, Forootan, FS, and Ke, Y. Increased Expression of Id1 and Id3 Promotes Tumorigenicity by Enhancing Angiogenesis and Suppressing Apoptosis in Small Cell Lung Cancer. Genes Cancer (2014) 5(5-6):212–25. doi:10.18632/genesandcancer.20
311. Sharma, BK, Kolhe, R, Black, SM, Keller, JR, Mivechi, NF, and Satyanarayana, A. Inhibitor of Differentiation 1 Transcription Factor Promotes Metabolic Reprogramming in Hepatocellular Carcinoma Cells. FASEB j (2016) 30(1):262–75. doi:10.1096/fj.15-277749
312. Singh, B, Berry, JA, Shoher, A, Ayers, GD, Wei, C, and Lucci, A. COX-2 Involvement in Breast Cancer Metastasis to Bone. Oncogene (2007) 26(26):3789–96. doi:10.1038/sj.onc.1210154
313. Baldwin, W, McRae, S, Marek, G, Wymer, D, Pannu, V, Baylis, C, et al. Hyperuricemia as a Mediator of the Proinflammatory Endocrine Imbalance in the Adipose Tissue in a Murine Model of the Metabolic Syndrome. Diabetes (2011) 60(4):1258–69. doi:10.2337/db10-0916
314. Gandhi, J, Khera, L, Gaur, N, Paul, C, and Kaul, R. Role of Modulator of Inflammation Cyclooxygenase-2 in Gammaherpesvirus Mediated Tumorigenesis. Front Microbiol (2017) 8:538. doi:10.3389/fmicb.2017.00538
315. Hou, Z., Falcone, D. J., Subbaramaiah, K., and Dannenberg, A. J.. Macrophages induce COX-2 expression in breast cancer cells: role of IL-1β autoamplification. Carcin (2011) 32(5):695–702. doi:10.1093/carcin/bgr027
316. Martínez-Reyes, C. P., Manjarrez-Reyna, A. N., Méndez-García, L. A., Aguayo-Guerrero, J. A., Aguirre-Sierra, B., Villalobos-Molina, R, et al. Uric Acid Has Direct Proinflammatory Effects on Human Macrophages by Increasing Proinflammatory Mediators and Bacterial Phagocytosis Probably via URAT1. Biomolecules (2020) 10:576. doi:10.3390/biom10040576
317. Lee, HE, Lee, JY, Yang, G, Kang, HC, Cho, Y-Y, Lee, HS, et al. Inhibition of NLRP3 Inflammasome in Tumor Microenvironment Leads to Suppression of Metastatic Potential of Cancer Cells. Scientific Rep (2019) 9(1):12277. doi:10.1038/s41598-019-48794-x
318. Bent, R, Moll, L, Grabbe, S, and Bros, M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Ijms (2018) 19(8):2155. doi:10.3390/ijms19082155
Keywords: cell cycle, adipokines, mesenchymal transition, leptin signaling, obesity derived cancers
Citation: Umar MI, Hassan W, Murtaza G, Buabeid M, Arafa E, Irfan HM, Asmawi MZ and Huang X (2021) The Adipokine Component in the Molecular Regulation of Cancer Cell Survival, Proliferation and Metastasis. Pathol. Oncol. Res. 27:1609828. doi: 10.3389/pore.2021.1609828
Received: 01 April 2021; Accepted: 09 July 2021;
Published: 13 September 2021.
Edited by:
Anna Sebestyén, Semmelweis University, HungaryCopyright © 2021 Umar, Hassan, Murtaza, Buabeid, Arafa, Irfan, Asmawi and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ghulam Murtaza, Z21kb2dhcjM1NkBnbWFpbC5jb20=; Manal Buabeid, bS5idWFiZWlkQGFqbWFuLmFjLmFl