 Linghong Kong1†
Linghong Kong1† Lining Wang
Lining Wang- 1Department of Pathology, Beijing Chuiyangliu Hospital, Beijing, China
- 2Hepato-Pancreato-Biliary Center, Beijing Tsinghua Changgung Hospital, School of Clinical Medicine, Tsinghua University, Beijing, China
Background: Few overlaps between prognostic biomarkers are observed among different independently performed genomic studies of esophageal squamous cell carcinoma (ESCC). One of the reasons for this is the insufficient cohort size. How many cases are needed to prognostic genes analysis in ESCC?
Methods: Here, based on 387 stage II/III ESCC cases analyzed by whole-genome sequencing from one single center, effects of cohort size on prognostic genes analysis were investigated. Prognostic genes analysis was performed in 100 replicates at each cohort size level using a random resampling method.
Results: The number of prognostic genes followed a power-law increase with cohort size in ESCC patients with stage II and stage III, with exponents of 2.27 and 2.25, respectively. Power-law curves with increasing events number were also observed in stage II and III ESCC, respectively, and they almost overlapped. The probability of obtaining statistically significant prognostic genes shows a logistic cumulative distribution function with respect to cohort size. To achieve a 100% probability of obtaining statistically significant prognostic genes, the minimum cohort sizes required in stage II and III ESCC were approximately 95 and 60, respectively, corresponding to a number of outcome events of 33 and 36, respectively.
Conclusion: In summary, the number of prognostic genes follows a power-law growth with the cohort size or events number in ESCC. The minimum events number required to achieve a 100% probability of obtaining a statistically significant prognostic gene is approximately 35.
Background
Esophageal squamous cell carcinoma (ESCC) is the most common histologic subtype of esophageal cancer and characterized by a high degree of clinical and genetic heterogeneity [1–3]. A reliable set of prognostic genes will contribute to a better understanding of the molecular mechanisms of ESCC progression and is crucial to guide clinical management. With the development of high-throughput sequencing technology, whole-exome sequencing (WES) or whole-genome sequencing (WGS) has been widely used for prognostic markers analysis in ESCC [4–11]. Over 1,000 ESCC exomes have been sequenced in the past years, however, little overlap between prognostic genes has been seen in the different ESCC studies [4–11]. The most straightforward explanation for this phenomenon is usually attributed to the fact that the cohorts used in different studies differed in certain potentially relevant factors (such as stage, gender, and genetic context). However, sample size is also an important influencing factor [12].
Somatic mutations have been detected in the coding regions of approximately 14,000 genes in ESCC, of which 65 genes showed mutation frequency of >5% [8]. In prognostic survival analysis, the number of outcome events should be sufficient relative to predictors [13]. For the identification of prognosis-related genes, the cohort size should be larger than the number of mutated genes. However, the cohort size for different genomic studies in ESCC is usually in the tens to hundreds. In addition, the number of outcome events is also an important factor for prognostic genes analysis. The statistical power of survival analysis actually depends on the number of outcome events rather than the total cohort size [14, 15].
Here, our aim is to define how many cases are needed to identify prognosis-related genes in ESCC? To exclude other influencing factors, such as genetic context, follow-up time, and staging, we have focused here on a single ESCC dataset from one center and investigated the effect of cohort size on prognostic genes using random resampling methods.
Methods
Study data
Somatic mutation data and clinical information of ESCC cases were obtained from the published study [8]. Of the patients, a total of 222 ESCC patients with stage II and 165 ESCC patients with stage III from one center had overall survival (OS) data and were selected for further analysis. The median follow-up time for stage II and III patients was 34 and 27 months, respectively. The number of OS outcome events for stage II and III patients was 77 and 99, respectively.
Prognostic genes analysis
Random resampling was performed by randomly selecting n cases in the dataset of stage II patients and the dataset of stage III patients, respectively. The cohort size n ranged from 1/7%–95% of all cases, with each cohort size being randomly sampled 100 times. Predictive analyses for prognostic genes associated with OS were also repeatedly performed 100 times for each cohort size level using the “maftools” package [16]. Survival analyses were determined using the Kaplan-Meier method and compared by the log-rank test. Differential genes with p < 0.05 were considered significant.
Statistical analysis
Statistical analysis was carried out using SPSS 22.0 software (SPSS, Inc., Chicago, IL, United States) or R statistical software (v4.1.0; R Core Team 2021). All results were presented as means ± standard error.
Results
In patients with stage II ESCC, the number of statistically significant prognostic genes increased with cohort size in a power-law with an exponent of 2.27 (Figure 1A). The power-law growth curve was also observed in patients with stage III ESCC and was generally consistent with the growth exponent of patients with stage II ESCC (Figure 1B). We further analyzed the relationship between the number of prognostic genes and the number of outcome events. Power-law growth curves were observed again for stage II and stage III ESCC, respectively, and they almost overlapped (Figure 1C). We then simulated and analyzed the relationship between the number of prognostic genes and the events number by mixing data from stage II and stage III ESCC. The best-fit curve also conformed to the power-law growth with R2 of 0.9917 (Figure 1D).
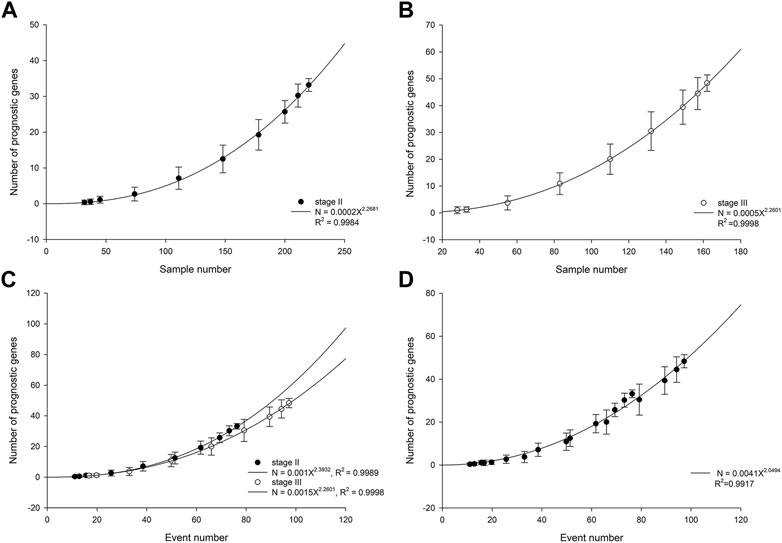
FIGURE 1. Number of prognostic genes increases with cohort size in stage II ESCC (A) and stage III ESCC (B). The lines are best-fit results for power-law growth N = 0.0002X2.2681 in stage II ESCC and N = 0.0005X2.2601 in stage III ESCC, respectively. (C) Number of prognostic genes increases with events number. The lines are best-fit results for power-law growth N = 0.001X2.3932 in stage II ESCC and N = 0.0015X2.2601 in stage III ESCC, respectively. (D) Mixing data from stage II and stage III, simulated number of prognostic genes increases with events number. The line is best-fit result for power-law growth N = 0.0041X2.0494.
We then analyzed the probability of obtaining at least one statistically significant prognostic gene in relation to cohort size. Logistic cumulative distribution curves were observed in both stage II and stage III ESCC patients (Figure 2). To achieve a 90% probability of obtaining statistically at least one statistically significant prognostic gene, the minimum cohort sizes required for stage II and III ESCC were approximately 65 and 45, respectively (Figure 2), which correspond to a number of events of 23 and 27, respectively. To achieve a 100% probability of obtaining at least one statistically significant prognostic gene, the minimum cohort sizes required for stage II and III ESCC were approximately 95 and 60, respectively (Figure 2), which correspond to a number of events of 33 and 36, respectively.
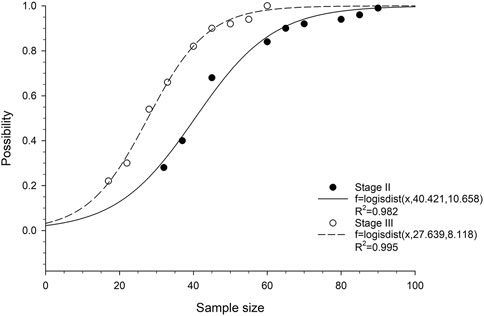
FIGURE 2. Correlation between probability of obtaining at least one prognostic gene and cohort size in ESCC patients. The fitted curves are based on logistic cumulative distribution functions.
Discussion
Prognosis is one of the core principles of medical practice. A number of studies have been conducted on the prediction of OS in ESCC based on WGS or WES [4–11]. However, few studies investigate the cohort size needed for prognostic genes studies. The prognostic genes identified by different studies showed very little overlap [4–11]. One reason is the inadequate cohort size. In this paper, we focused on a single ESCC dataset from one center and investigated the effect of cohort size on prognostic genes using random resampling methods. This cohort is the largest ESCC cohort to date from a single clinical center and includes 437 ESCC cases from Han population of Shanxi, China [8]. This cohort nicely excludes the interference of genetic background differences. A total of 387 patients with stage II/III ESCC were enrolled in the study after verification of clinical information.
In both stage II and stage III ESCC patients, the number of prognostic genes showed a power-law relationship with increasing cohort size, although the specific parameters of the formula differed. However, prognostic analysis is based on time-to-event data [17]. The events number is more critical than cohort size in prognostic analysis. Relative to the events number, the growth curves of the number of prognostic genes in patients with stage II and stage III ESCC largely overlapped. Although the cohort sizes required for prognostic genes analysis of stage II and stage III ESCC are different, the number of outcome events required is essentially the same. These results indicated that the power-law growth of the number of prognostic genes with cohort size is common in ESCC, independent of stage.
Prognostic studies are usually retrospective and cohort sizes are rarely considered prior to analysis [18]. However, the prognostic genes obtained on limited data are nothing but misleading. Our results showed that at least 35 outcome events are required in ESCC to ensure the acquisition of statistically significant prognostic genes.
A limitation of this study is that the number of ESCC patients from one center (99 outcome events) is still insufficient, resulting in the plateau in the number of prognostic genes not reached. Enrolling more patients will detect more mutated genes, thereby increasing the number of prognostic genes. However, theoretically there should be a plateau in the number of prognostic genes. The number of cases or events needed to reach this plateau still needs to be further explored.
Conclusion
In summary, the number of prognostic genes takes a power-law growth with cohort size in ESCC. Our results suggest that at least 35 outcome events are required for genomic mutation-based prognostic studies in ESCC. These results will help to the trial design of prognostic genes analysis in ESCC.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The original data analyzed in this study are publicly available and come from the published study (PMID: 32398863). Ethical review and approval were not required for the study on human participants in accordance with the local legislation and institutional requirements. This study did not require informed consent for participation in accordance with the national legislation and institutional requirements.
Author contributions
LW conceived the study and reviewed the manuscript. LK, MY, and ZW contributed to data collection and statistical analysis. LK and MY drafted the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lam, AK. Molecular biology of esophageal squamous cell carcinoma. Crit Rev Oncol Hematol (2000) 33:71–90. doi:10.1016/s1040-8428(99)00054-2
2. Arnal, MJD, Arenas, ÁF, and Arbeloa, ÁL. Esophageal cancer: Risk factors, screening and endoscopic treatment in Western and Eastern countries. World J Gastroenterol (2015) 21:7933–43. doi:10.3748/wjg.v21.i26.7933
3. Lin, DC, Wang, MR, and Koeffler, HP. Genomic and epigenomic aberrations in esophageal squamous cell carcinoma and implications for patients. Gastroenterology (2018) 154:374–89. doi:10.1053/j.gastro.2017.06.066
4. Lin, DC, Hao, JJ, Nagata, Y, Xu, L, Shang, L, Meng, X, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet (2014) 46:467–73. doi:10.1038/ng.2935
5. Gao, YB, Chen, ZL, Li, JG, Hu, XD, Shi, XJ, Sun, ZM, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet (2014) 46:1097–102. doi:10.1038/ng.3076
6.The Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature (2017) 541:169–75. doi:10.1038/nature20805
7. Moody, S, Senkin, S, Islam, SMA, Wang, J, Nasrollahzadeh, D, Cortez Cardoso Penha, R, et al. Mutational signatures in esophageal squamous cell carcinoma from eight countries with varying incidence. Nat Genet (2021) 53:1553–63. doi:10.1038/s41588-021-00928-6
8. Cui, Y, Chen, H, Xi, R, Cui, H, Zhao, Y, Xu, E, et al. Whole-genome sequencing of 508 patients identifies key molecular features associated with poor prognosis in esophageal squamous cell carcinoma. Cell Res (2020) 30:902–13. doi:10.1038/s41422-020-0333-6
9. Song, Y, Li, L, Ou, Y, Gao, Z, Li, E, Li, X, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature (2014) 509:91–5. doi:10.1038/nature13176
10. Sawada, G, Niida, A, Uchi, R, Hirata, H, Shimamura, T, Suzuki, Y, et al. Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology (2016) 150:1171–82. doi:10.1053/j.gastro.2016.01.035
11. Zhang, N, Shi, J, Shi, X, Chen, W, and Liu, J. Mutational characterization and potential prognostic biomarkers of Chinese patients with esophageal squamous cell carcinoma. Onco Targets Ther (2020) 13:12797–809. doi:10.2147/OTT.S275688
12. Ein-Dor, L, Kela, I, Getz, G, Givol, D, and Domany, E. Outcome signature genes in breast cancer: Is there a unique set? Bioinformatics (2005) 21:171–8. doi:10.1093/bioinformatics/bth469
13. Riley, RD, Snell, KI, Ensor, J, Burke, DL, Harrell, FE, Moons, KG, et al. Minimum sample size for developing a multivariable prediction model: PART II - binary and time-to-event outcomes. Stat Med (2019) 38:1276–96. doi:10.1002/sim.7992
14. Schober, P, and Vetter, TR. Survival analysis and interpretation of time-to-event data: The tortoise and the hare. Anesth Analg (2018) 127:792–8. doi:10.1213/ANE.0000000000003653
15. In, J, and Lee, DK. Survival analysis: Part II - applied clinical data analysis. Korean J Anesthesiol (2019) 72:441–57. doi:10.4097/kja.19183
16. Mayakonda, A, Lin, DC, Assenov, Y, Plass, C, and Koeffler, HP. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res (2018) 28:1747–56. doi:10.1101/gr.239244.118
17. Moons, KG, Royston, P, Vergouwe, Y, Grobbee, DE, and Altman, DG. Prognosis and prognostic research: What, why, and how? Bmj (2009) 338:b375. doi:10.1136/bmj.b375
Keywords: esophageal squamous cell carcinoma, prognostic genes analysis, power-law, events number, cohort size
Citation: Kong L, Yang M, Wan Z and Wang L (2023) Cohort size required for prognostic genes analysis of stage II/III esophageal squamous cell carcinoma. Pathol. Oncol. Res. 29:1610909. doi: 10.3389/pore.2023.1610909
Received: 30 October 2022; Accepted: 26 January 2023;
Published: 07 February 2023.
Edited by:
Andrea Ladányi, National Institute of Oncology (NIO), HungaryReviewed by:
Tamás Micsik, Semmelweis University, HungaryJános Tibor Fekete, Semmelweis University, Hungary
Copyright © 2023 Kong, Yang, Wan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lining Wang, d2FuZ2xpbmluZzEwMDFAMTI2LmNvbQ==
†These authors have contributed equally to this work