 Anna L. Kiss
Anna L. Kiss- Department of Anatomy, Histology and Embryology, Semmelweis University, Budapest, Hungary
The inflammation is an important biological response induced by various harmful stimuli, like viruses, bacterial infections, toxins, toxic compounds, tissue injury. During inflammation inflammatory cytokines and reactive oxygen species are produced. Inflammatory cytokines act on various receptors present on the plasma membrane of target cells. To initiate signaling cascade, and activate transcription factors, receptors should be internalized and enter the early endosomes, where the members of the signaling cascade can meet. The further cytoplasmic fate of the receptor plays crucial role in the progression and the course of inflammation. Usually acute inflammation removes injurious stimuli and helps to regain the normal healthy status of the organism. In contrast to this the uncontrolled chronic inflammation—stimulating other than immune cells, inducing transdifferentiation—can provide base of various serious diseases. This paper draws the attention of the long-lasting consequence of chronic inflammation, pointing out that one of the most important step in medication is to identify in time the factors initiating and maintaining inflammation.
Inflammation
The inflammation is a defense process of the body, a biological response of the immune system to harmful stimuli. The inflammation can be triggered by various pathogens (viruses, bacteria) toxins, toxic compounds, tissue injury [1]. These harmful stimuli initiate a chemical signaling cascade, activating leukocytes, that then produce and release inflammatory cytokines [2], such as interleukin-1β (IL-1β), interleukin-6, tumor necrosis factor-α (TNF-α). These cytokines interact with and activate receptors (IL-6R, TNFR-1, TNFR-2, TLR4, GM-CSFR etc.) [3]. Receptor activation triggers the phosphorylation of various signaling molecules such as mitogen-activated protein kinase (MAPK), nuclear factor kappa-B (NF-kB), Janus kinase (Jak), resulting in the activation of various transcription factors. This coordinate activation of signaling molecules regulates the level of inflammatory mediators in resident tissue cells, and recruit inflammatory cells from the blood [4,5]. Thus the acute inflammation is a protective mechanism, removes the injurious stimuli and initiates a healing process, restoring the homeostasis of the organism [6].
Uncontrolled acute inflammation, however, can become chronic, and can provide the base of a variety of serious, chronic diseases (tumors, a variety of neurogenerative diseases like Alzheimer disease, Parkinson disease, multiple sclerosis, lateral sclerosis, autoimmune diseases, diabetes, cardiovascular diseases, fibrosis etc.) [7–9]. Although the pathogenesis of these diseases is different, in most cases the inflammatory mediators, the regulatory and signaling pathways are common. In all cases 1) receptors must be present on the cell membrane and should be internalized (to register and transmit signals through the plasma membrane into the cell interior); 2) the members of the signaling cascade (various kinases, transcription factors) should meet in a platform called “signaling organelles/endosomes” [10], where they become activated; 3) activated transcription factors have to be translocated into the nucleus, to regulate inflammatory genes, resulting in inflammatory cytokine synthesis and release into the environment.
Reactive Oxygen Species and Inflammation
Reactive oxygen species (ROS) are side-products of normal cell metabolism and produced in various cellular compartments like endoplasmic reticulum, mitochondria, peroxisomes. These reduced metabolites, with their strong oxidative capabilities, oxidize proteins, lipids, cellular constituents and can cause serious DNA damage. In physiological concentration ROS act as second messengers and function as signaling molecules in cell growth, cell adhesion and cell differentiation [11,12]. As second messengers, ROS posttranslationally modify proteins by oxidizing redox-sensitive cystein residues [13]. An increasing number of evidence show that reactive oxygen species are involved in initiation, progression and resolution of inflammatory responses. Chronic inflammation results in increasing ROS production. In turn, ROS regulate various types of kinases and transcription factors, such as nuclear kappa B (NF-kB), that are involved in the activation of pro-inflammatory genes [14,15]. Overproduction of ROS during chronic inflammation results in cell and tissue injury driving to serious diseases [13,16]. Neutrophil granulocytes are the primary ROS producers in the immune system, but other phagocytic cells, like macrophages, are also able to produce ROS significantly contributing to the increased ROS level. During post-bacterial phagocytosis NADPH oxidase (NOX) bound to the phagosomal membrane is activated and produces superoxide [17]. Altered metabolic activity, oxidative stress can also induce ROS production. Inflammatory cytokines activate STAT3, that is translocated into the nucleus, and acts as a transcription factor, regulating the transcription of inflammatory genes [18]. Recently it was shown, that a pool of Ser727 phosphorylated STAT3 translocates into the mitochondria where it stimulates ROS production [19–22], proving that inflammatory cytokines can directly stimulate ROS production. During chronic inflammation the ROS and inflammatory cytokine production are most probably interactive, orchestrated and synchronized and they magnify each other’s effect.
Inflammation-Induced Epithelial-to-Mesenchymal Transition
Nowadays it is well known that not only immune cells are able to respond to inflammatory stimuli and produce inflammatory cytokines. Under special circumstances, such as inflammatory stimuli, epithelial cells undergo a transition called epithelial-to-mesenchymal transition (EMT). EMT is an important biological process, triggered by various extracellular signals, including cytokines, growth factors and extracellular matrix components [23]. Three types of EMT are known, one of them, the EMT type II is associated with inflammation, wound healing, tissue regeneration and organ fibrosis [24]. It has been demonstrated that one of the most important factor that initiate EMT type II is the transforming growth factor (TGFβ) [25]. During this inflammation-induced EMT, epithelial cells undergo a complex proteomic remodelling, a series of morphological and biochemical changes [26]. In our in vivo system (rat mesenteric mesothelial cells) we found that inflammatory stimuli (Freund’s adjuvant induced peritonitis) led to prominent, spectacular morphological and biochemical changes in these epithelial cells. These mesothelial cells lost their polarity, E-cadherin and β-catenin synthesis was down-regulated, as a result of this cellular junctions were disassembled [23,27]. The basement membrane degraded, the cytoskeleton was remodelled and reorganized [27]. The volume of the cytoplasm, the number of the cytoplasmic organelles (rough ER, mitochondria, Golgi vesicles) dramatically increased, indicating that the metabolic rate and synthetic activities of these cells highly increased, but autophagy was blocked [28,29]. We have shown that healthy, non-treated mesothelial cells express toll-like receptor 4 (TLR4) as well (our unpublished results), that is responsible to recognize pathogens and participate in the activation of the inflammatory stimulus [9]. As a result of inflammation, mesothelial cells start to express macrophage markers (OX43, ED1) [30,31], synthesize and secrete pro-inflammatory cytokines, TNFα, IL-6 [32,33], their phagocytic activity is highly increased [32]. Inflammatory stimuli also initiate the expression and nuclear translocation of an unique transcription factor, EGR1 [32], which is known to affect the monocyte/macrophage lineage [34–37]. All these data provide strong evidence that under inflammatory stimuli, as a result of EMT, mesothelial cells transdifferentiate into macrophages or macrophage-like cells. Granulocyte-macrophage-colony stimulating factor (GM-CSF) was originally defined as a member of the hematopoietic cytokine family, promoting the survival and activation of granulocytes, macrophages and dendritic cells differentiation in vivo [32]. In our in vitro system (mesentery culture), GM-CSF treatment resulted in exactly the same morphological and biochemical changes as Freund’s adjuvant treatment proving that GM-CSF itself induced EMT, having an inflammatory effect as well [32]. GM-CSF-treated mesothelial cells synthesize and release GM-CSF and express GM-CSF receptor (both α and β) as well, constituting an autocrine, self-stimulating system. These data strongly support the previous observations, that GM-CSF can be locally produced [38–40], and point out the importance of GM-CSF in promoting inflammation. Taking together all these data, we can conclude that parallel to the morphological alterations, the inflammatory cytokines activated epithelial cells (in our case mesothelial cells) themselves started to produce pro-inflammatory cytokines, expressed cytokine receptors on their plasma membrane constituting an autocrine, self-stimulating, self-generating system. Thus these cells can contribute, actively take part in the inflammatory response (Figure 1).
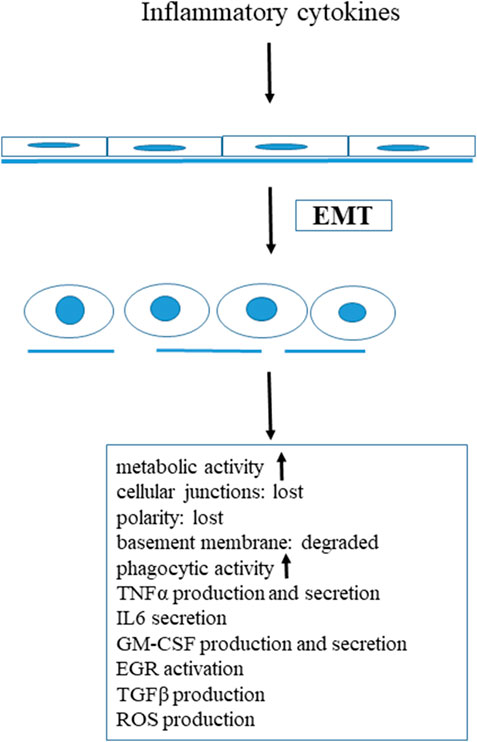
FIGURE 1. Inflammatory stimuli induced transdifferentiation of mesenteric mesothelial cells.
Receptors: Key Players in Response to Inflammatory Stimuli
Extracellular stimuli inducing inflammatory response are hydrophilic molecules that require specific receptors on the plasma membrane of the target cell to transmit the signal into the cell interior where further steps of the signal transduction (transcription factors activation) take place. Thus membrane receptors are key elements of the inflammatory response, their presence on the plasma membrane and their intracellular fate determine how far the inflammatory response lasts, and how strong it is. Most of the cytokine receptors consist of several chains associated with different non-receptor kinases. Ligand binding to the receptors (TLR4, GM-CSFR, TNFR-1, and 2 etc.) induces synchronized and orchestrated phosphorylation events, initiating a signaling cascade (MAPK, NF-kB, JAK, STAT pathways) in the cell [41–44]. For the interaction between downstream molecules and initiating efficient signaling, membrane receptors, with their cargo, have to be internalized via clathrin-coated vesicles, caveolae (highly hydrophobic caveolin-1 containing plasma membrane invaginations), or lipid rafts [45,46]. After pinching off these vesicles from the plasma membrane, they fuse with early endosomes or “signaling” endosomes, (also called signaling organelles) [45,47–49]. Early or “signaling” endosomes provide a platform, a “meeting room” for the interactions with the down-stream elements of various signaling cascades (“signaling crosstalk”) [10]. After initiating signal, the intracellular route of the receptor and its fate plays a crucial role in the inflammatory response. If the receptor recycles to the cell surface, another signaling cycle can start, and the inflammatory stimulus is being continuously transmitted to the cell interior. Alternatively, if the cell directs the receptor to the degradative (lysosomal) pathway, the inflammation is stopped, and the cell can start to regenerate itself. In our experimental system we have shown that for the efficient signal transduction the receptor internalization is essential. When blocking the caveola-mediated internalization of GM-CSF receptor β by dynasore, (a potent inhibitor of small GTP-ase, dynamin [50]), the downstream element of GM-CSF signaling cascade, the STAT5, was not phosphorylated [46]. We also found that at the initial period of inflammation the receptor was present in early endosomes, where the STAT5 phosphorylation most probably occurs. While the inflammatory reaction was in course, the receptor was detected in recycling endosomes. When mesenteric mesothelial cells started to regenerate, the receptor was found in Rab7 positive late endosomes, and directed to a degradative/lysosomal pathway [46]. These results clearly show that removing the receptors from the cell surface, the inflammation stops and the regeneration can start, proving that the intracellular fate of the receptors should crucially determine the course of the inflammation (Figure 2).
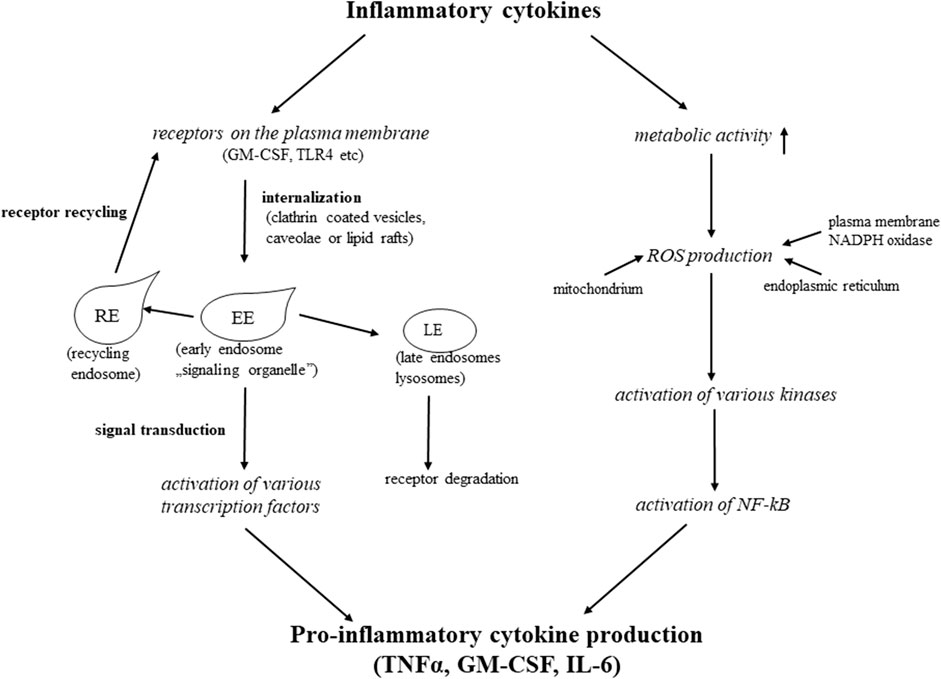
FIGURE 2. Mechanisms of inflammatory cytokines actions.
Clinical Aspects
Nowadays it is more and more accepted that chronic inflammation provides the base of serious chronic, non-inflammatory diseases including cardiovascular, neurodegenerative and bowel diseases, diabetes, arthritis, fibrosis and cancer. In the aetiology of the Alzheimer’s disease scientists found links between various pathogens and the development of the disease [51–53]. There are data indicating that in lateral sclerosis and in multiple sclerosis bacterial or viral infection can be one of the factors that initiates the disease. In normal healthy circumstances the inflammatory mediators are expressed at a low level and have only a few effects on the central nervous system. Neurons, astrocytes, microglial cells and oligodendrocytes can produce inflammatory mediators, and cytokine receptors are also expressed in the central nervous system [54]. Most probably these cytokines and receptors contribute to the normal physiological function of the CNS [55]. In pathological conditions, however, bacterial, viral, and fungi infections as well as tissue injuries, the pro-inflammatory cytokine level is highly elevated, partly because they are produced by the microglial cells and the recruited macrophages at the site of injury. Cytokines can also be produced at the periphery, at sites of infections or injuries, and transported by the blood. They can cross the blood-brain barrier by active transport through the leaky region of the vascular endothelial layer. These blood-derived cytokines can significantly contribute to the increased level of inflammatory mediators in the CNS. Various forms of IL-1, TNFα, IL-6, together with free radicals, ROS (causing damage of lipids, proteins and DNA) can significantly promote myelin sheath loss, injury of neurons, inducing neurodegeneration, serious neurodegenerative diseases like Alzheimer’s disease, lateral sclerosis, multiple sclerosis, Parkinson disease [56].
Chronic inflammation often results in fibrosis of various organs. Following injury, fibrosis plays an essential role in tissue repair. The aim of this process is to produce and deposit connective tissue components (fibers, and other extracellular matrix components), to regenerate the original tissue architecture [57]. Myofibroblasts are the main connective tissue components producing cells. Under inflammatory stimuli they derive from fibroblasts, macrophages, endothelial cells and many other cells types as well [57]. Injury results in local inflammation, and the inflammatory cytokines stimulate the transformation of the above mentioned cells into myofibroblasts. The long-lasting inflammatory stimuli, the uncontrolled chronic inflammation, however, can magnify the amplitudo of the production and deposition of the connective tissue components, resulting in chronic fibrosis in various organs (heart, kidney, liver etc).
Inflammation is far more complex process than it was thought originally. Under inflammatory stimuli (pathogens, toxins, tissue injuries etc.) inflammatory cytokines and ROS are produced primarily by immune cells and are transported by the blood circulation transporting them to everywhere in the body. Thus they act not only locally, instead their effect is rather systemic. An increasing number of evidence support that other cells, including epithelial cells, can respond to inflammatory stimuli, and can contribute to chronic inflammation. As it was discussed before mesenteric mesothelial cells are also equipped, or under inflammatory stimuli, can be equipped with all the tools necessary for inflammatory responses [28,29,31–33]. Under inflammatory stimuli they undergo transdifferentiation and as a result of this transformation they become able to produce inflammatory cytokines and ROS. With this capability they join to the immune defend “army” of the body and amplify, multiply and enhance the inflammatory response.
New data are making our knowledge deeper and wider and the “story” is becoming more and more complex. Recently it has been demonstrated that obesity is also associated with chronic systemic inflammation and is a serious risk factor for development of breast, ovarian and endometrial cancers [57–59]. Adipocytes produce adipokines and steroid hormones, like estrogen. Obesity alters the production of these molecules resulting in an inflammatory microenvironment in which pro-inflammatory macrophages can produce inflammatory cytokines. Additionally, free fatty acids are released from the adipocytes in obese individuals stimulating toll-like receptor 4 (TLR4) and activating NF-kB signaling pathway. The latter upregulates secretion of TNFα, IL-6, IL-8, IL-1β creating a positive feed-forward loop to sustain chronic inflammation and cancer progression [60–63].
Recently the so-called post-Covid diseases draw the attention to the generalized effect of inflammation. During SARS-COV-2 virus infection not only the immune cells but—as a result of inflammation induced EMT—epithelial cells in the lung (alveolar pneumocytes, endothelial cells and pleural mesothelial cells) can also produce high amounts of cytokines, resulting in a “cytokine storm.” Since inflammatory cytokines induce EMT, the transdifferentiation of various cells (including epithelial cells present in the blood vessels, in the lung alveoli and in the pleura) into myofibroblast can easily occur. Myofibroblasts then produce large amount of connective tissue fibers and other components of the extracellular matrix, locally resulting in lung fibrosis [64]. If we consider that the inflammatory cytokines are transported by the blood everywhere in the body, they can affect all the organs of the body, and depending on the individual’s sensitivity, they can induce various chronic diseases, causing cardiovascular, neural and mental symptoms.
Conclusion
Since chronic inflammation can provide the base of many serious, fatal diseases, in the preventive therapy to identify pathogens, viruses, bacteria inducing inflammation is crucial. Because of the long-lasting consequence of chronic inflammation (inducing serious diseases) the most critical steps in medication are to identify, recognize inflammation in time, to determine the factors (bacteria, viruses, tissue injuries) that are behind the inflammation and use the proper treatment. The treatment should be combined using antibiotics, anti-inflammatory and anti-oxidant drugs.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to express my great thanks to Professor Pál Röhlich for the precious comments and accurate language correction of the manuscript. I am also very much grateful for Viktória Zsiros for her technical help.
References
1. Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell (2010) 140:771–6. doi:10.1016/j.cell.2010.03.006
2. Jabbour, HN, Sales, KJ, Catalano, RD, and Norman, JE. Inflammatory Pathways in Female Reproductive Health and Disease. Reprod (2009) 138:903–19. doi:10.1530/rep-09-0247
3. Kaminska, B. MAPK Signalling Pathways as Molecular Targets for Anti-inflammatory Therapy-From Molecular Mechanisms to Therapeutic Benefits. Biochim Biophys Acta (Bba) - Proteins Proteomics (2005) 1754:253–62. doi:10.1016/j.bbapap.2005.08.017
4. Lawrence, T. The Nuclear Factor NF-kB Pathway in Inflammation. CSH Perspect Biol (2009) 1:a001651. doi:10.1101/cshperspect.a001651
5. Libby, P. Inflammatory Mechanisms: the Molecular Basis of Inflammation and Disease. Nutr Rev (2007) 65:S140–6. doi:10.1111/j.1753-4887.2007.tb00352.x
6. Ferrero-Miliani, L, Nielsen, OH, Andersen, PS, and Girardin, SE. Chronic Inflammation: Importance of NOD2 and NALP3 in Interleukin-1beta Generation. Clin Exp Immunol (2007) 147:227–35. doi:10.1111/j.1365-2249.2006.03261.x
7. Nathan, C, and Ding, A. Nonresolving Inflammation. Cell (2010) 140:871–82. doi:10.1016/j.cell.2010.02.029
8. Zhou, Y, Hong, Y, and Huang, H. Triptolide Attenuates Inflammatory Response in Membranous Glomerulo-Nephritis Rat via Downregulation of NF-Κb Signaling Pathway. Kidney Blood Press Res (2016) 41:901–10. doi:10.1159/000452591
9. Chen, L, Deng, H, Cui, H, Fang, J, Zuo, Z, Deng, J, et al. Inflammatory Responses and Inflammation-Associated Diseases in Organs. Oncotarget (2018) 9:7204–18. doi:10.18632/oncotarget.23208
10. Pálfy, M, Reményi, A, and Korcsmáros, T. Endosomal Crosstalk: Meeting Points for Signaling Pathways. Trends Cel Biol (2012) 22:447–56. doi:10.1016/j.tcb.2012.06.004
11. Tichá, T, Lochman, J, Činčalová, L, Luhová, L, and Petřivalský, M. Redox Regulation of Plant S-Nitrosoglutathione Reductase Activity through post-translational Modifications of Cysteine Residues. Biochem Biophysical Res Commun (2017) 494:27–33. doi:10.1016/j.bbrc.2017.10.090
12. Thannickai, VJ, and Fanburg, BL. Reactive Oxygen Spesies in Cell Signaling. Am J Physiol Lung Cel Mol Physiol (2000) 279:1005–28.
13. Chelomstitko, MA. Role of Reactive Oxygen Species in Inflammation: a Minireview. Mosc Univ Biol Sci Bull (2018) 73:199–202. doi:10.3103/S009639251804003X
14. Flohé, L, Brigelius-Flohé, R, Saliou, C, Traber, MG, and Packer, L. Redox Regulation of NF-Kappa B Activation. Free Radic Biol Med (1997) 22:1115–26. doi:10.1016/s0891-5849(96)00501-1
15. Ranneh, Y, Ali, F, Akim, AM, Hamid, HA, Khazaai, H, and Fadel, A. Crosstalk between Reactive Oxygen Species and Pro-inflammatory Markers in Developing Various Chronic Diseases: a Review. Appl Biol Chem (2017) 60(3):327–38. doi:10.1007/s13765-017-0285-9
16. Henríquez-Olguín, C, Altamirano, F, Valladares, D, López, JR, Allen, PD, and Jaimovich, E. Altered ROS Production, NF-Κb Activation and Interleukin-6 Gene Expression Induced by Electrical Stimulation in Dystrophic Mdx Skeletal Muscle Cells. Biochim Biophys Acta (Bba) - Mol Basis Dis (2015) 1852:1410–9. doi:10.1016/j.bbadis.2015.03.012
17. Leto, TL, and Geiszt, M. Role of NOX Family NADPH Oxidases in Host Defense. Antioxid Redox Signal (2006) 8(9-10):1549–61. doi:10.1089/ars.2006.8.1549
18. Walker, JG, and Smith, MD. The Jak-STAT Pathway in Rheumatoid Arthritis. J Rheumatol (2005) 32:1650–3.
19. Balic, JJ, Albargy, H, Luu, K, Kirby, FJ, Jayasekara, WSN, Mansell, F, et al. STAT3 Serine Phosphorylation Is Required for TLR4 Metabolic Reprogramming and IL-1β Expression. Nat Commun (2020) 11:3816. doi:10.1038/s41467-020-17669-5
20. Wegrzyn, J, Potla, R, Chwae, Y-J, Sepuri, NBV, Zhang, Q, Koeck, T, et al. Function of Mitochondrial Stat3 in Cellular Respiration. Science (2009) 323:793–7. doi:10.1126/science.1164551
21. Gough, DJ, Corlett, A, Schlessinger, K, Wegrzyn, J, Larner, AC, and Levy, DE. Mitochondrial STAT3 Supports Ras-dependent Oncogenic Transformation. Science (2009) 324:1713–6. doi:10.1126/science.1171721
22. Gough, DJ, Marié, IJ, Lobry, C, Aifantis, I, and Levy, DE. STAT3 Supports Experimental K-RasG12D-Induced Murine Myeloproliferative Neoplasms Dependent on Serine Phosphorylation. Blood (2014) 124:2252–61. doi:10.1182/blood-2013-02-484196
23. Boyer, B, Vallés, AM, and Edme, N. Induction and Regulation of Epithelial-Mesenchymal Transitions. Biochem Pharmacol (2000) 60:1091–9. doi:10.1016/s0006-2952(00)00427-5
24. Lee, JM, Dedhar, S, Kalluri, R, and Thompson, EW. The Epithelial-Mesenchymal Transition: New Insights in Signaling, Development, and Disease. J Cel Biol (2006) 172:973–81. doi:10.1083/jcb.200601018
25. Strutz, F, Zeisberg, M, Ziyadeh, FN, Yang, C-Q, Kalluri, R, Müller, GA, et al. Role of Basic Fibroblast Growth Factor-2 in Epithelial-Mesenchymal Transformation. Kidney Int (2002) 61:1714–28. doi:10.1046/j.1523-1755.2002.00333.x
26. Kalluri, R, and Weinberg, RA. The Basics of Epithelial-Mesenchymal Transition. J Clin Invest (2009) 119:1420–8. doi:10.1172/jci39104
27. Savagner, P. Leaving the Neighborhood: Molecular Mechanisms Involved during Epithelial-Mesenchymal Transition. BioEssays (2001) 23:912–23. doi:10.1002/bies.1132
28. Zsiros, V, and Kiss, AL. Cellular and Molecular Events of Inflammation-Induced Transdifferentiation (EMT) and Regeneration (MET) in Mesenteric Mesothelial Cells. Inflamm Res (2020) 69(12):1173–9. doi:10.1007/s00011-020-01400-7
29. Zsiros, V, Katz, S, Dóczi, N, and Kiss, AL. Autophagy Is the Key Process in the Re-establishment of the Epitheloid Phenotype during Mesenchymal-Epithelial Transition (MET). Exp Cel Res (2017) 352:382–92. doi:10.1016/j.yexcr.2017.02.031
30. Katz, S, Balogh, P, and Kiss, AL. Mesothelial Cells Can Detach from the Mesentery and Differentiate into Macrophage-like Cells. APMIS (2011) 119:782–93. doi:10.1111/j.1600-0463.2011.02803.x
31. Katz, S, Balogh, P, Nagy, N, and Kiss, AL. Epithelial-To-Mesenchymal Transition Induced by Freund's Adjuvant Treatment in Rat Mesothelial Cells: A Morphological and Immunocytochemical Study. Pathol Oncol Res (2012) 18:641–9. doi:10.1007/s12253-011-9489-1
32. Katz, S, Zsiros, V, Dóczi, N, and Kiss, AL. Inflammation-Induced Epithelial-To-Mesenchymal Transition and GM-CSF Treatment Stimulate Mesenteric Mesothelial Cells to Transdifferentiate into Macrophages. Inflammation (2018) 41:1825–34. doi:10.1007/s10753-018-0825-4
33. Katz, S, Zsiros, V, and Kiss, AL. Under Inflammatory Stimuli Mesenteric Mesothelial Cells Transdifferentiate into Macrophages and Produce Pro-inflammatory Cytokine IL-6. Inflamm Res (2019) 68:525–8. doi:10.1007/s00011-019-01247-7
34. Laslo, P, Spooner, CJ, Warmflash, A, Lancki, DW, Lee, H-J, Sciammas, R, et al. Multilineage Transcriptional Priming and Determination of Alternate Hematopoietic Cell Fates. Cell (2006) 126:755–66. doi:10.1016/j.cell.2006.06.052
35. Krishnaraju, K, Nguyen, HQ, Liebermann, DA, and Hoffman, B. The Zinc finger Transcription Factor Egr-1 Potentiates Macrophage Differentiation of Hematopoietic Cells. Mol Cel Biol (1995) 15:5499–507. doi:10.1128/mcb.15.10.5499
36. Krishnaraju, K, Hoffman, B, and Liebermann, DA. The Zinc finger Transcription Factor Egr-1 Activates Macrophage Differentiation in M1 Myeloblastic Leukemia Cells. Blood (1998) 92:1957–66. doi:10.1182/blood.v92.6.1957
37. Krishnaraju, K, Hoffman, B, and Liebermann, DA. Early Growth Response Gene 1 Stimulates Development of Hematopoietic Progenitor Cells along the Macrophage Lineage at the Expense of the Granulocyte and Erythroid Lineages. Blood (2001) 97:1298–305. doi:10.1182/blood.v97.5.1298
38. Kay, AB, Ying, S, Varney, V, Gaga, M, Durham, SR, Moqbel, R, et al. Messenger RNA Expression of the Cytokine Gene Cluster, Interleukin 3 (IL-3), IL-4, IL-5, and Granulocyte/macrophage colony-stimulating Factor, in Allergen-Induced Late-phase Cutaneous Reactions in Atopic Subjects. J Exp Med (1991) 173:775–8. doi:10.1084/jem.173.3.775
39. Al-Saffar, N, Khwaja, HA, Kadoya, Y, and Revell, PA. Assessment of the Role of GM-CSF in the Cellular Transformation and the Development of Erosive Lesions Around Orthopaedic Implants. Am J Clin Pathol (1996) 105:628–39. doi:10.1093/ajcp/105.5.628
40. Carrieri, PB, Provitera, V, Rosa, TD, Tartaglia, G, Gorga, F, and Perrella, O. Profile of Cerebrospinal Fluid and Serum Cytokines in Patients with Relapsing-Remitting Multiple Sclerosis. A Correlation with Clinical Activity. Immunopharmacology and Immunotoxicology (1998) 20:373–82. doi:10.3109/08923979809034820
41. Johnson, JA, Bacon, CM, Riedy, MC, and O1Shea, JJ. Signaling by IL-2 and Related Cytokines: JAKs, STATs and Relationship to Immunodeficiency. J Leukocyt Biol (1996) 60:441–52.
42. Campbell, PM, and Halloran, PF. T Cell Activation. In: NL Tilney, TB Strom, and LC Paul, editors. Transplantation Biology: Cellular and Molecular Aspects. Philadelphia: Lippicott-Raven (1996). p. 411–34.
43. Hendrayani, S-F, Al-Harbi, B, Al-Ansari, MM, Silva, G, and Aboussekhra, A. The Inflammatory/cancer-Related IL-6/STAT3/NF-κB Positive Feedback Loop Includes AUF1 and Maintains the Active State of Breast Myofibroblasts. Oncotarget (2016) 7:41974–85. doi:10.18632/oncotarget.9633
44. Kyriakis, JM, and Avruch, J. Mammalian Mitogen-Activated Protein Kinase Signal Transduction Pathways Activated by Stress and Inflammation. Physiol Rev (2001) 81:807–69. doi:10.1152/physrev.2001.81.2.807
45. Le Roy, C, and Wrana, JL. Clathrin- and Non-clathrin-mediated Endocytic Regulation of Cell Signalling. Nat Rev Mol Cel Biol (2005) 6:112–26. doi:10.1038/nrm1571
46. Zsiros, V, Katz, S, Doczi, N, and Kiss, AL. Endocytosis of GM-CSF Receptor β Is Essential for Signal Transduction Regulating Mesothelial-Macrophage Transition. Biochim Biophys Acta (Bba) - Mol Cel Res (2019) 1866:1450–62. doi:10.1016/j.bbamcr.2019.06.005
47. Miaczynska, M, Pelkmans, L, and Zerial, M. Not just a Sink: Endosomes in Control of Signal Transduction. Curr Opin Cel Biol (2004) 16:400–6. doi:10.1016/j.ceb.2004.06.005
48. Howe, CL, and Mobley, WC. Signaling Endosome Hypothesis: A Cellular Mechanism for Long Distance Communication. J Neurobiol (2004) 58:207–16. doi:10.1002/neu.10323
49. Polo, S, and Di Fiore, PP. Endocytosis Conducts the Cell Signaling Orchestra. Cell (2006) 124:897–900. doi:10.1016/j.cell.2006.02.025
50. Macia, E, Ehrlich, M, Massol, R, Boucrot, E, Brunner, C, and Kirchhausen, T. Dynasore, a Cell-Permeable Inhibitor of Dynamin. Develop Cel (2006) 10:839–50. doi:10.1016/j.devcel.2006.04.002
51. Carrosco, L, Pisa, D, and Alonso, R. Polymicrobial Infections and Neurodegeneration. Curr Clin Micro Rpt (2020) 7:20–30. doi:10.1007/s40588-020-00139-3
52. Miklossy, J. Chronic Inflammation and Amyloidogenesis in Alzheimer's Disease - Role of Spirochetes1. Jad (2008) 13:381–91. doi:10.3233/jad-2008-13404
53. Vigasova, D, Nemergut, M, Liskova, B, and Damborsky, J. Multi-pathogen Infections and Alzheimer's Disease. Microb Cel Fact (2021) 20:25. doi:10.1186/s12934-021-01520-7
54. Allan, SM, and Rothwell, NJ. Inflammation in central Nervous System Injury. Phil Trans R Soc Lond B (2003) 358:1669–77. doi:10.1098/rstb.2003.1358
55. Pollmacher, T, Haack, M, Schuld, A, Reichenberg, A, and Yirmiya, R. Low Levels of Circulating Inflammatory Cytokines-Do They Affect Human Brain Functions? Brain Behav Immun (2002) 16:525–32. doi:10.1016/s0889-1591(02)00004-1
56. Lucas, S-M, Rothwell, NJ, and Gibson, RM. The Role of Inflammation in CNS Injury and Disease. Br J Pharmacol (2006) 147:S232–S240. doi:10.1038/sj.bjp.0706400
57. Ueha, S, Shand, FHW, and Matsushima, K. Cellular and Molecular Mechanisms of Chronic Inflammation-Associated Organ Fibrosis. Front Immun (2012) 3:71. doi:10.3389/fimmu.2012.00071
58. Deng, T, Lyon, CJ, Bergin, S, Caligiuri, MA, and Hsueh, WA. Obesity, Inflammation, and Cancer. Annu Rev Pathol Mech Dis (2016) 11:421–49. doi:10.1146/annurev-pathol-012615-044359
59. Picon-Ruiz, M, Pan, C, Drews-Elger, K, Jang, K, Besser, AH, Zhao, D, et al. Interactions between Adipocytes and Breast Cancer Cells Stimulate Cytokine Production and Drive Src/Sox2/miR-302b-Mediated Malignant Progression. Cancer Res (2016) 76:491–504. doi:10.1158/0008-5472.can-15-0927
60. Diaz-Ruano, A, Martinez-Alarcon, N, Ferrandiz-Cabrera, I, Marchal, JA, and Picon-Ruiz, M. Estron But Not Estradiol, Potentiates NF-kB-Driven Inflammation, EMT and Stemness in ER+epithelial Cells. Int J Mol Sci (2019) 20:528–47.
61. Howe, LR, Subbaramaiah, K, Hudis, CA, and Dannenberg, AJ. Molecular Pathways: Adipose Inflammation as a Mediator of Obesity-Associated Cancer. Clin Cancer Res (2013) 19:6074–83. doi:10.1158/1078-0432.ccr-12-2603
62. Picon-Ruiz, M, Morata-Tarifa, C, Valle-Goffin, JJ, Friedman, ER, and Slingerland, JM. Obesity and Adverse Breast Cancer Risk and Outcome: Mechanistic Insights and Strategies for Intervention. CA: A Cancer J Clinicians (2017) 67:378–97. doi:10.3322/caac.21405
63. Tornatore, L, Thotakura, AK, Bennett, J, Moretti, M, and Franzoso, G. The Nuclear Factor Kappa B Signaling Pathway: Integrating Metabolism with Inflammation. Trends Cel Biol (2012) 22:557–66. doi:10.1016/j.tcb.2012.08.001
Keywords: EMT, inflammatory cytokines, reactive oxygen species, receptor internalization, chronic inflammation
Citation: L. Kiss A (2022) Inflammation in Focus: The Beginning and the End. Pathol. Oncol. Res. 27:1610136. doi: 10.3389/pore.2021.1610136
Received: 19 October 2021; Accepted: 09 December 2021;
Published: 04 January 2022.
Edited by:
Anna Sebestyén, Semmelweis University, HungaryCopyright © 2022 L. Kiss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna L. Kiss, a2lzcy5hbm5hQG1lZC5zZW1tZWx3ZWlzLXVuaXYuaHU=